Abstract
Studies from the psychiatric genomics consortium have concluded that common schizophrenia risk variants are regulatory and have low effect sizes. Thus, one could either inherit many of these variants or have a dysfunction in an upstream mechanism that controls their expression; however, demonstration of the latter has remained elusive.
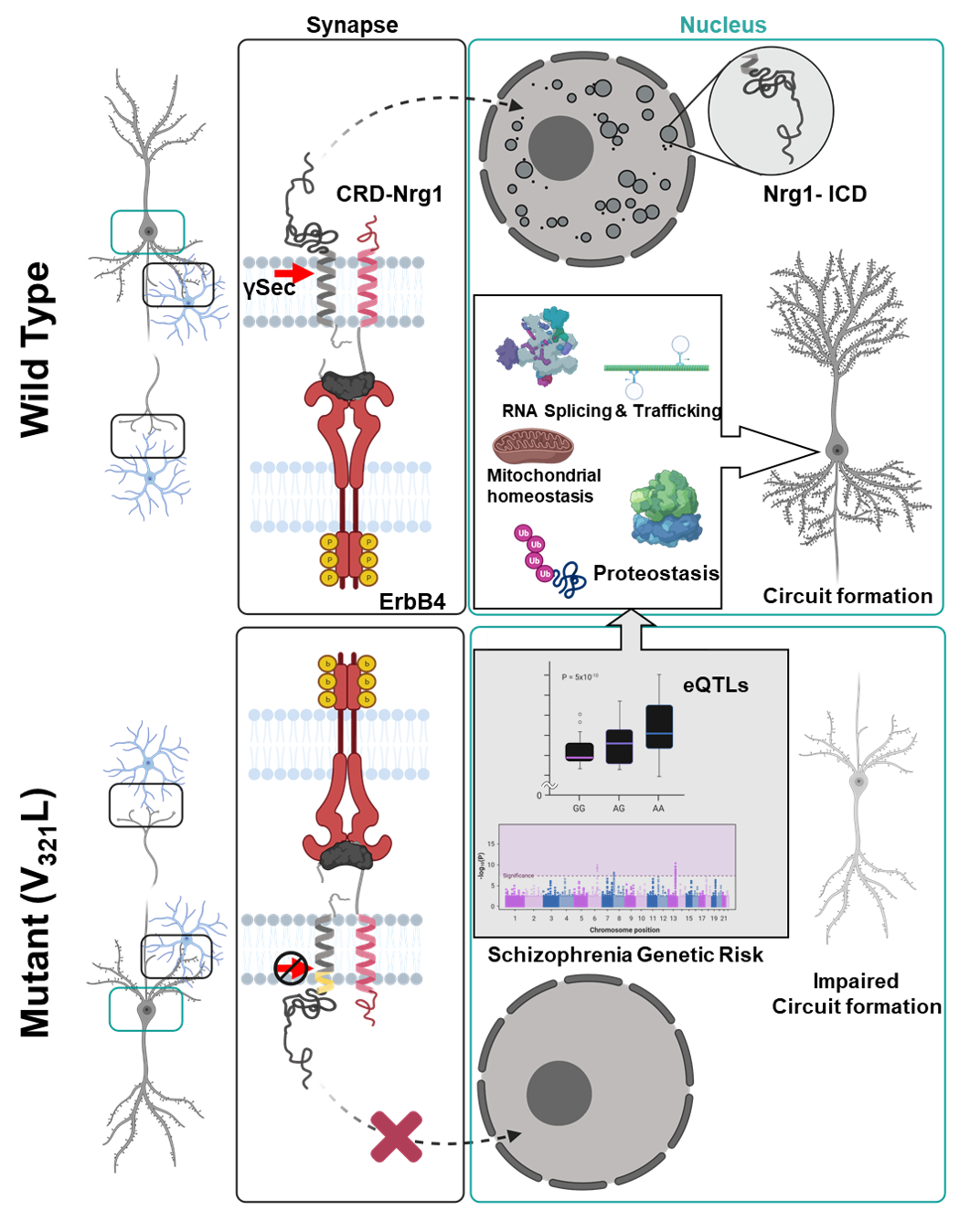
Neuregulin1 is a schizophrenia-susceptibility gene. Type III Neuregulin1 is cleaved by gamma secretase to produce a nucleus-targeted cytosolic fragment (ICD). A psychosis-associated mutation in Nrg1 (V321L) disrupts this cleavage implicating nuclear signaling in underlying pathology.
Using transcriptomic and network analyses, we uncovered a common gene regulatory network providing insights into the molecular pathology underlying genetic susceptibility to psychosis. Our data show that nuclear ICD regulates the expression of schizophrenia risk genes. The nuclear ICD signal originates at the synapse, hinting that expression of these risk genes is normally regulated during synaptogenesis; a process dysregulated in schizophrenia.