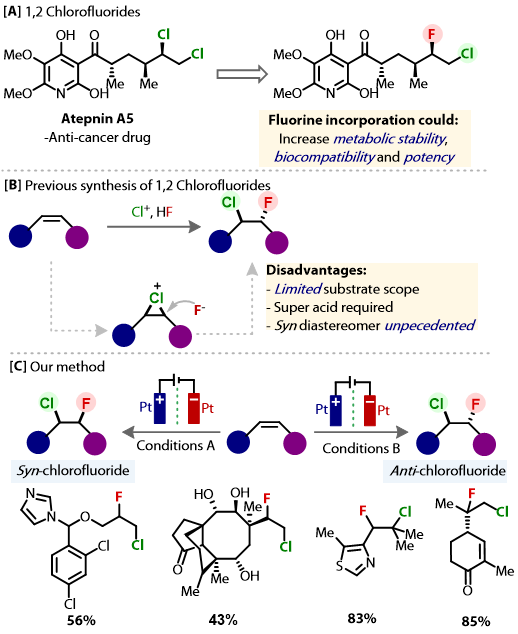
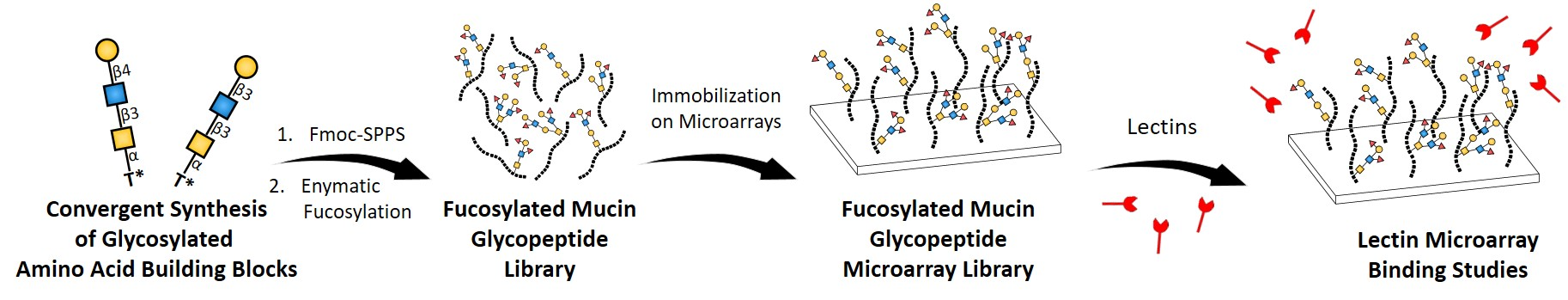
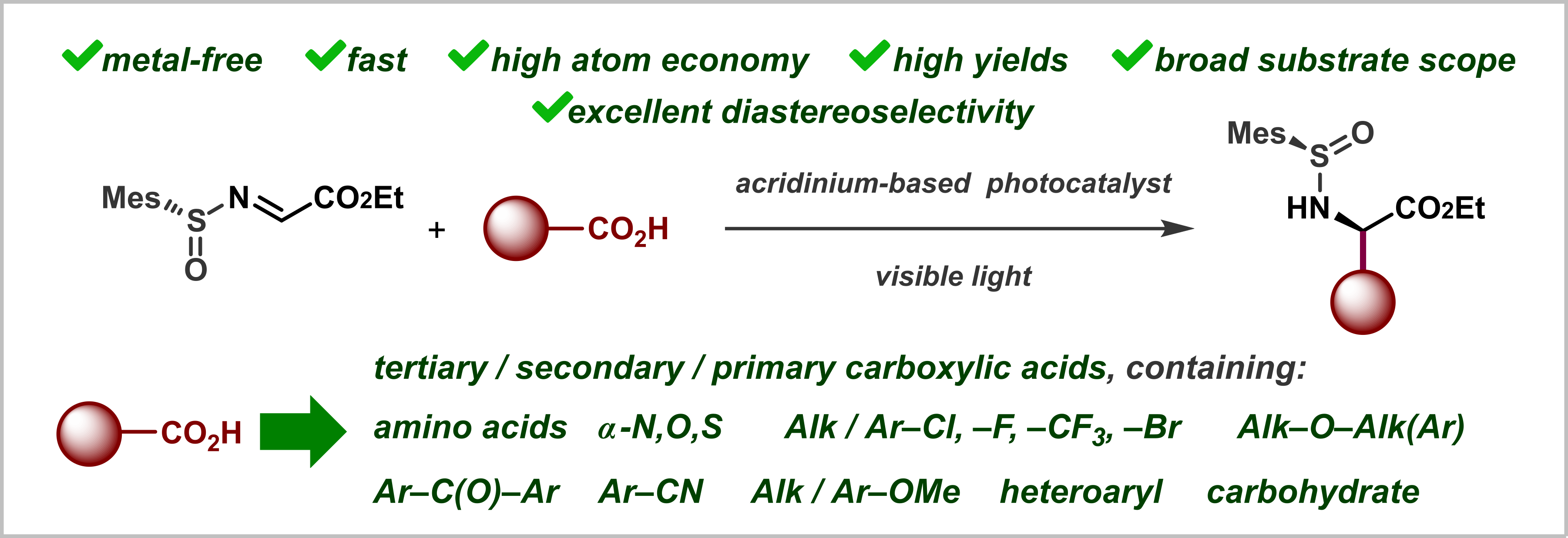
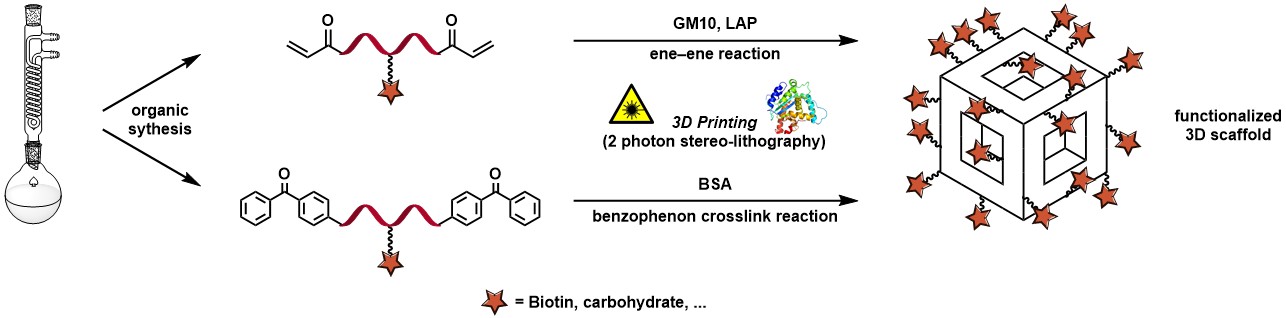
Abstract
The four Bromo and Extra-Terminal (BET) proteins, Brd2, Brd3, Brd4 and Brdt, play a crucial role in transcriptional regulation and other processes such as cell proliferation and inflammation. Each of the BET proteins contains two tandem bromodomains which are conserved across the family and are responsible for binding to acetylated lysine residues such as those at the N-terminus of histones. They have become an attractive therapeutic target as misregulation of BET proteins have been linked to diseases such as cancer, neurological disorders and inflammation.
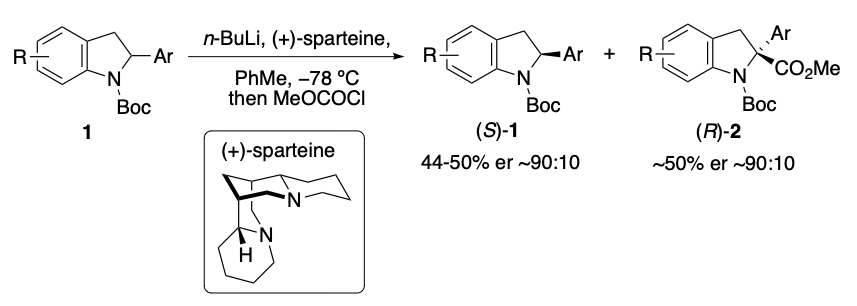
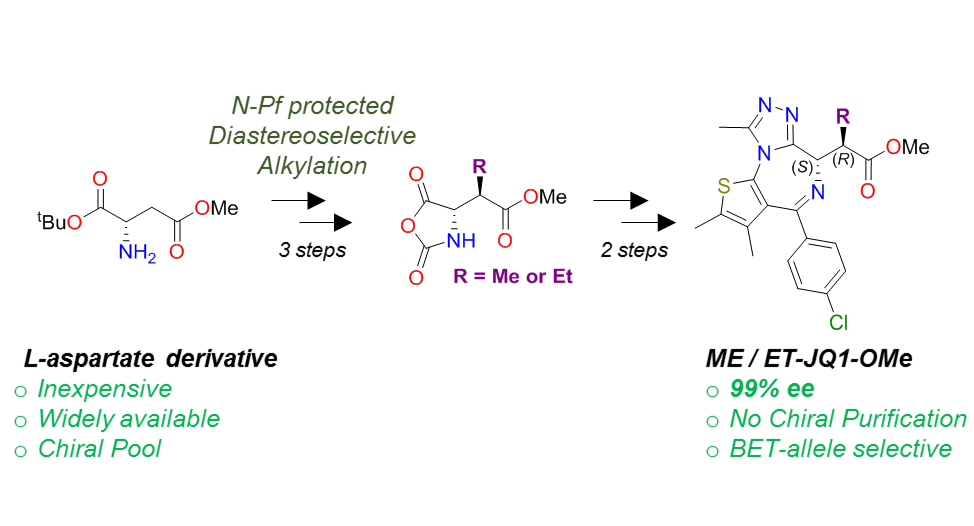
There are many examples of BET inhibitors, including JQ11 and I-BET762, 2 however, due to the high homology of BET bromodomains, these inhibitors are pan-selective and cannot discriminate between the bromodomains within the BET family. Our group has previously established a bump-&-hole approach to overcome this by selectively targeting a conserved Leucine residue found in BET bromodomains with alkylated I-BET762 derived probes ET3 and 9-ME-1.4 A limitation to the current synthetic route of these probes is that they require chiral separation following any alkylation step. Separation can be costly and lead to low yields due to loss of material.
We have achieved a new stereoselective route to obtain a key precursor which can be used in a divergent synthesis to yield a variety of bumped inhibitors containing a benzodiazepine scaffold. We demonstrate application of this new route to create novel bumped JQ1-analogues in >99% ee. Our new synthesis has also provided, for the first time, unambiguous evidence to the absolute stereochemistry of the active mutant-selective bumped ligand.
References
1. P. Filippakopoulos et al., Nature, 2010, 468, 1067.
2. O. Mirguet et al., J. Med. Chem., 2013, 56, 19, 7501-7515.
3. M. G. J. Baud et al., Science, 2014, 346, 638-641.
4. A. C. Runcie et al., Chem. Sci., 2018, 9, 2452-2468.