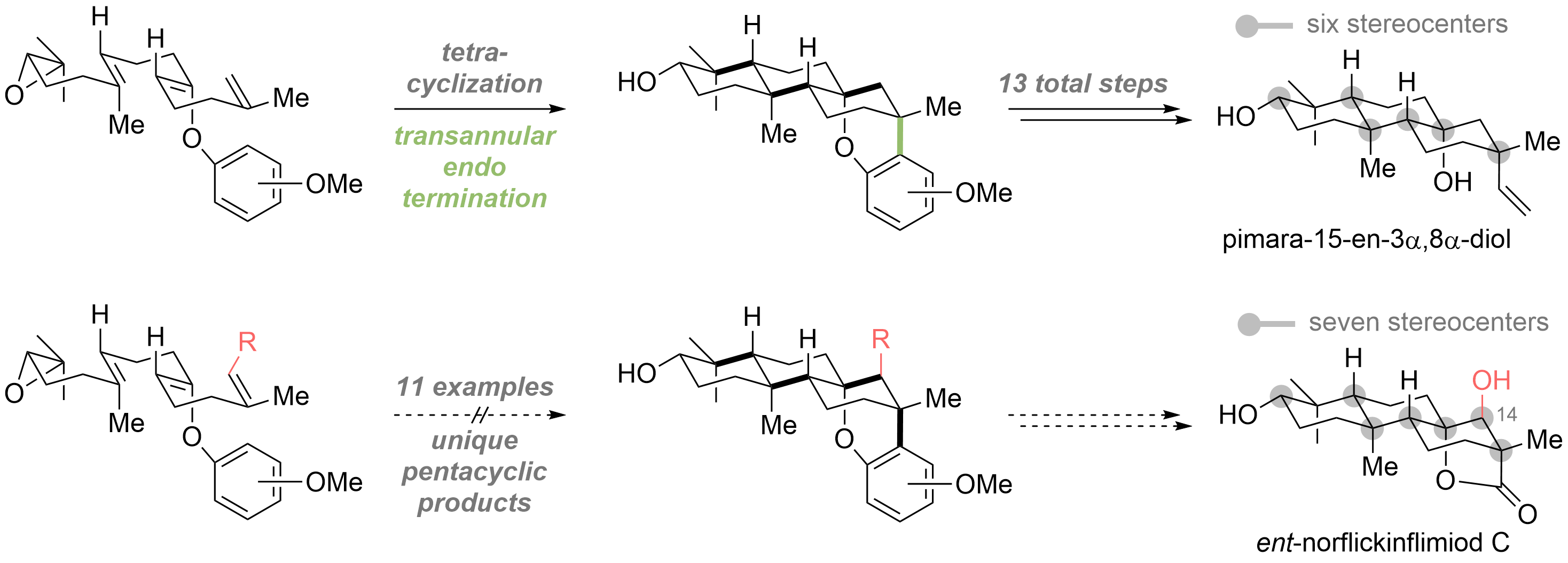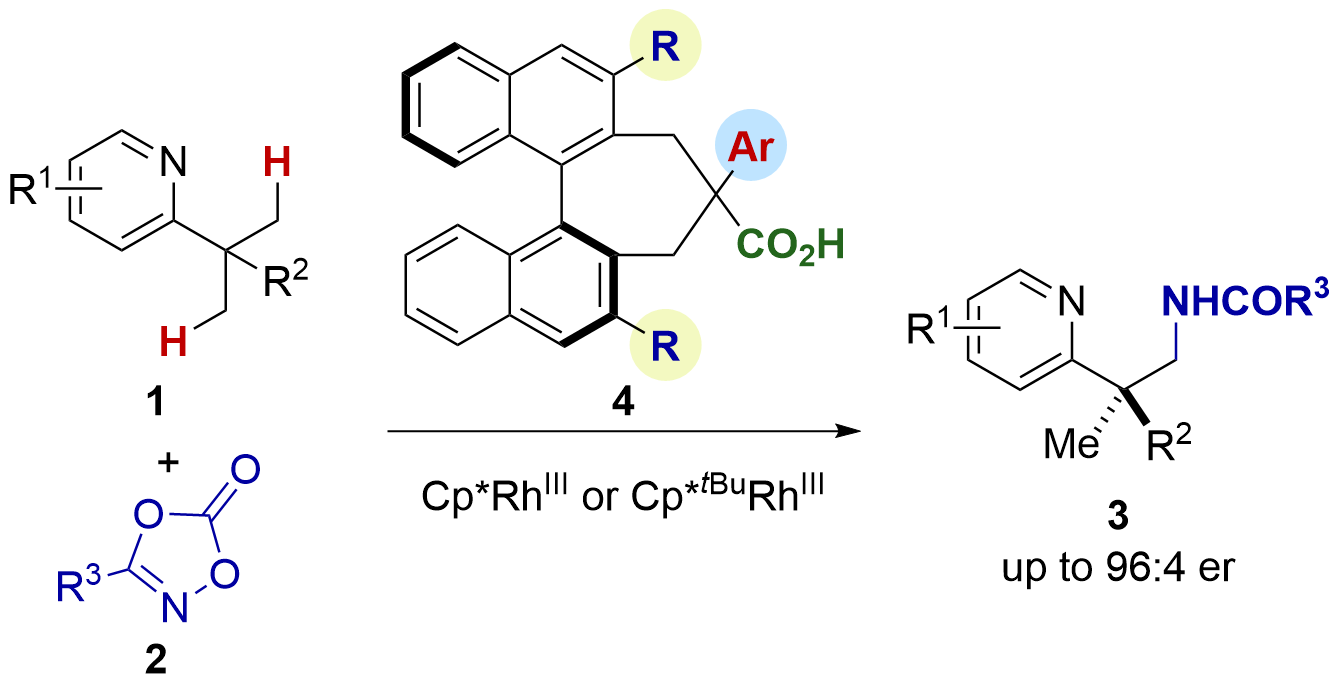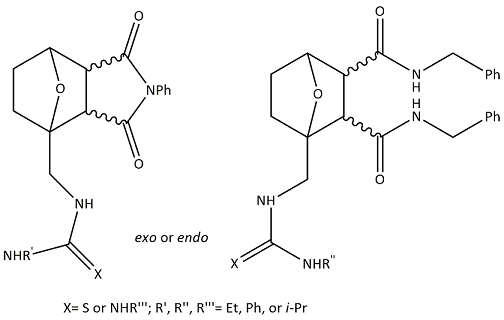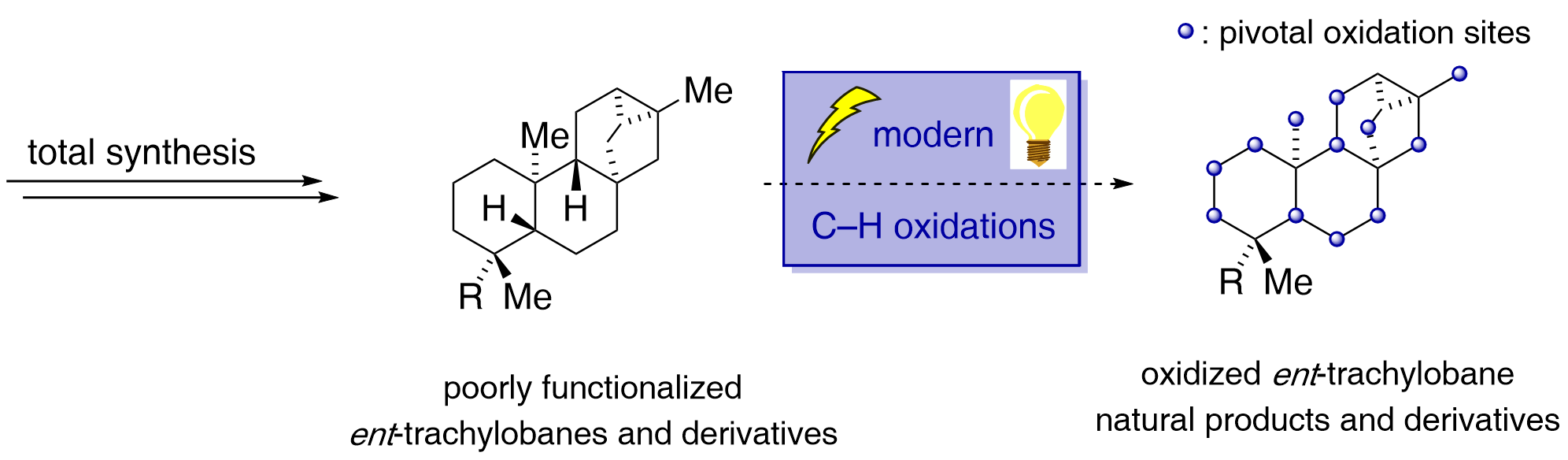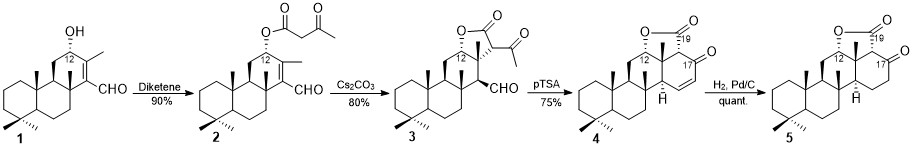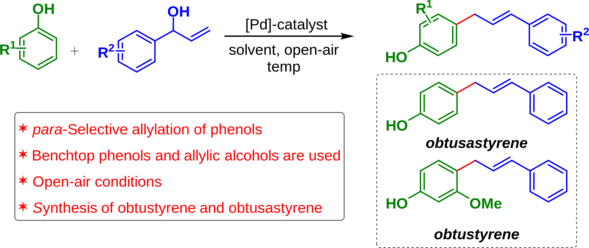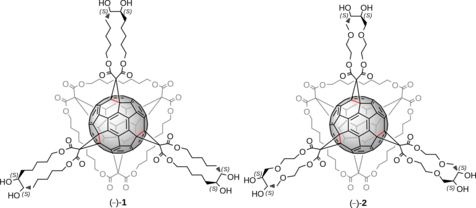
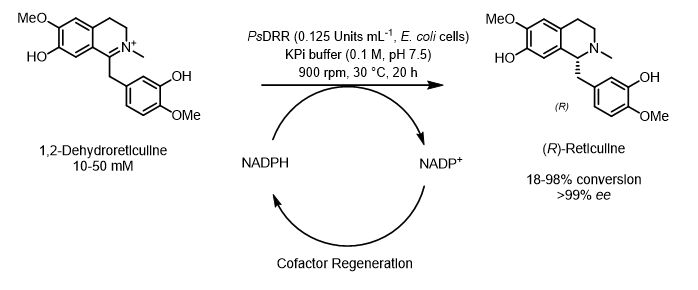
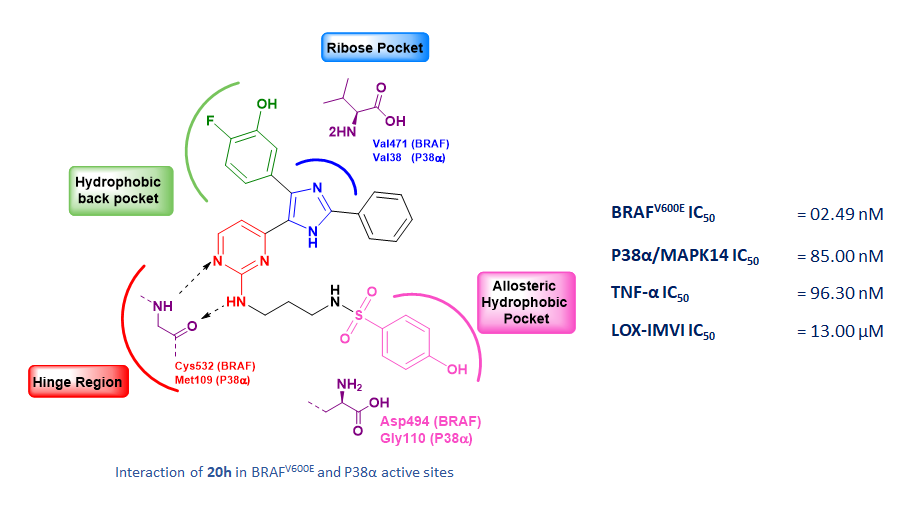
The emergence of giant shape amphiphiles
[1] synthesized by the modification of single molecular nanoparticles (MNPs)
[2] has set new perspectives in the area of supramolecular aggregation. C
60 has been defined as a MNP due to the rigid and highly symmetrical molecular structure. The prediction of the self-assembled architectures in relation to the exact molecular shape has not been realized yet setting the design and synthesis of new C
60 shape amphiphiles and the characterization of their nano-structures a primary target. Type I [3:3]-hexakis adducts of C
60[3] offer an excellent platform for designing a diversity of giant shape amphiphiles by incorporating different hydrophobic and hydrophilic addends in their structures. We designed and synthesized the giant shape amphiphiles (–)-
1 and (–)-
2 equipped with three
cyclo-monomalonate addends on one hemisphere of C
60 and a
cyclo-[3]-octylmalonate addend on the other. Each addend bears a 1,2-diol moiety which serves as the hydrophilic head. The structural difference between (–)-
1 and (–)-
2 originates from the different size and chemical nature of the
cyclo-monomalonates. Studies on their self-assembling behaviour by TEM and SEM revealed that the minor modification of their addends and consequently, the shape of these giant molecules, significantly influences their corresponding self-assembling architectures.
[4] The self-assembling behaviour of (–)-
1 in water revealed the remarkable ability of this giant shape amphiphile to form stable, giant vesicles with diameters varying from 500 nm up to 5
μm. The fullerene vesicles formed were found to be robust and retained their structural integrity in air or under vacuum, a behaviour inherent to giant molecules. However, the giant shape amphiphilic adduct (–)-
2 afforded mostly ill-defined, clustered, spherical aggregates with diameters ranging from 20 to 50 nm.
[1] W.-B. Zhang, X. Yu, C.-L. Wang, H.-J. Sun, I.-F. Hsieh, Y. Li, X.-H. Dong, K. Yue, R. Van Horn, S. Z. D. Cheng, Macromolecules 2014, 47, 1221.
[2] X. Yu, K. Yue, I.-F. Hsieh, Y. Li, X.-H. Dong, C. Liu, Y. Xin, H.-F. Wang, A.-C. Shi, G. R. Newkome, R.-M. Ho, E.-Q. Chen, W.-B. Zhang, S. Z. D. Cheng, Proc. Natl. Acad. Sci. 2013, 110, 10078.
[3] F. Djojo, E. Ravanelli, O. Vostrowsky, A. Hirsch, Eur. J. Org. Chem. 2000, 1051.
[4] C. P. Ioannou, G. I. Ioannou, E. E. Moushi, K. Velonia, N. Chronakis, Eur. J. Org. Chem., 2015, 4598.
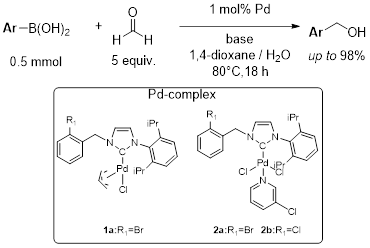
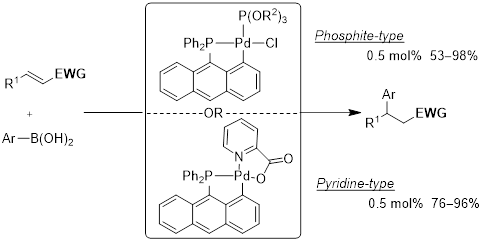
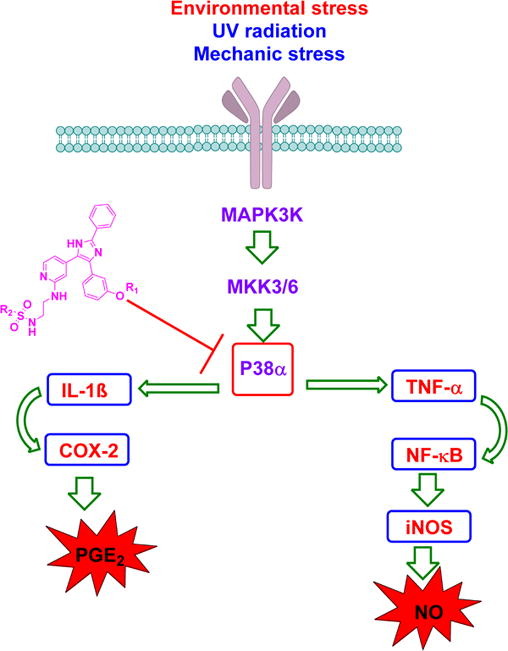
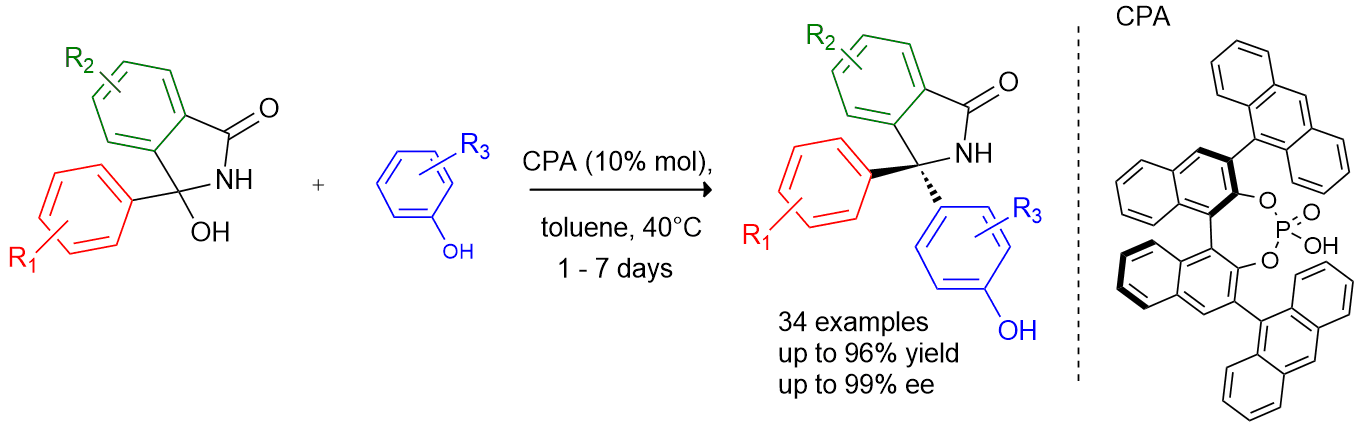
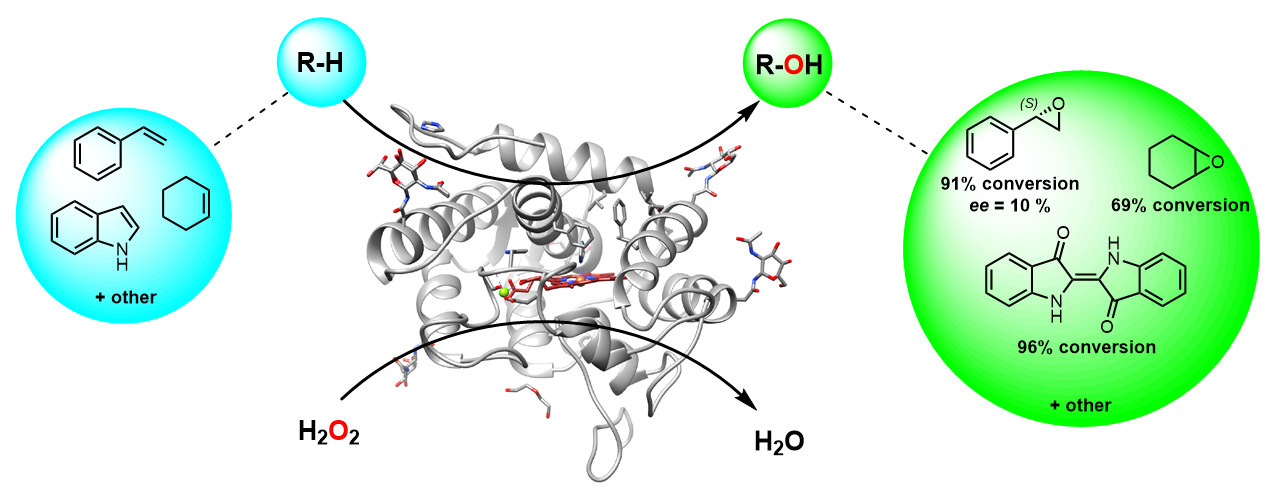
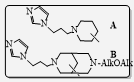
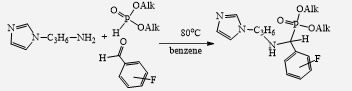
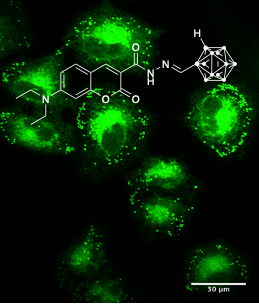
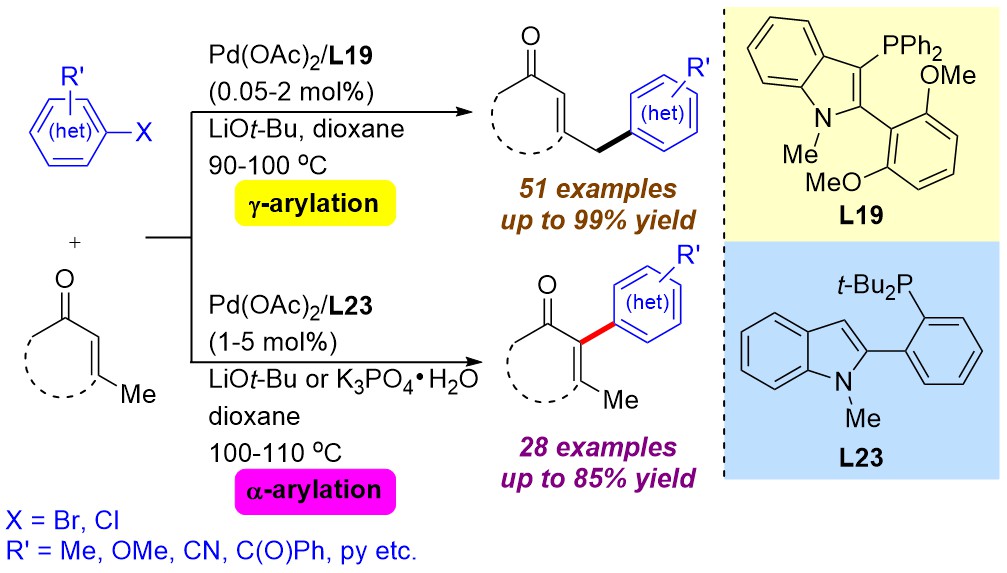

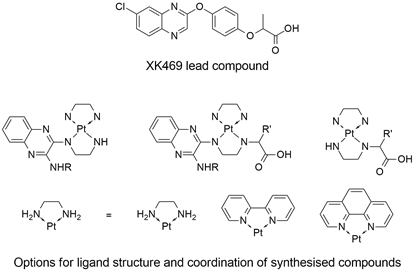
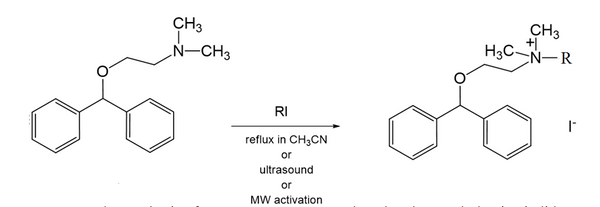
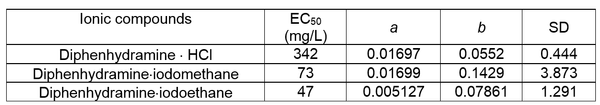

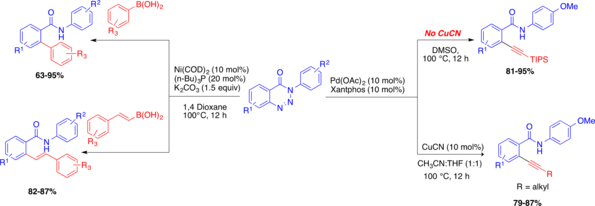
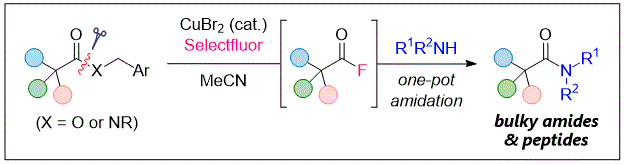
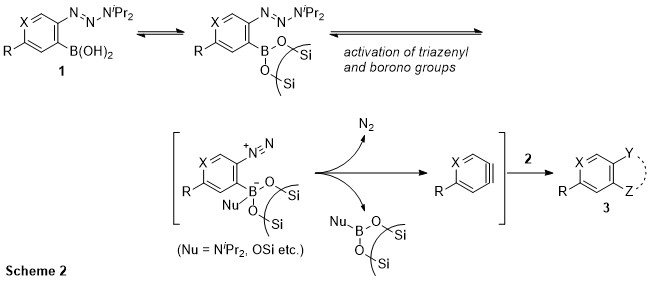
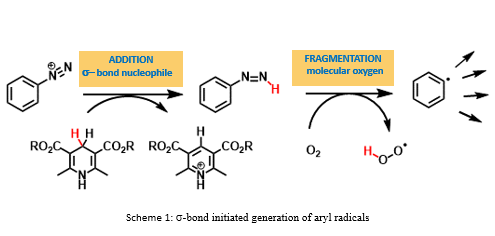 This presentation details a new approach for the metal-free reduction of aryl diazonium salts with Hantzsch esters as -bond nucleophiles (scheme 1).e This novel protocol leads to unprecedented operational simplicity: The reactions are very rapid and proceed in open air; there is no need for external irradiation or heating; and the process is compatible with many radical reactions. We illustrate these advantages by using the polar-radical crossover strategy in direct and cascade reactions by generated aryl radicals to regioselectively arylate a series of heterocyclic compounds; to synthesize ketones by arylation of silyl enol ethers; to synthesize benzothiophene and phenathrene derivatives by radical annulation reactions and effectively generate alkyl radicals in the cascade reactions from corresponding alkyl iodides to synthesize functionalized ketones, esters, sulfones and organophosphorus compounds from Michael acceptors. We demonstrated the uses of novel polar-radical-crossover cascade reaction in late stage functionalization of peptides, sugars, steroids, nucleosides and several representatives of antihistamine drugs.
This presentation details a new approach for the metal-free reduction of aryl diazonium salts with Hantzsch esters as -bond nucleophiles (scheme 1).e This novel protocol leads to unprecedented operational simplicity: The reactions are very rapid and proceed in open air; there is no need for external irradiation or heating; and the process is compatible with many radical reactions. We illustrate these advantages by using the polar-radical crossover strategy in direct and cascade reactions by generated aryl radicals to regioselectively arylate a series of heterocyclic compounds; to synthesize ketones by arylation of silyl enol ethers; to synthesize benzothiophene and phenathrene derivatives by radical annulation reactions and effectively generate alkyl radicals in the cascade reactions from corresponding alkyl iodides to synthesize functionalized ketones, esters, sulfones and organophosphorus compounds from Michael acceptors. We demonstrated the uses of novel polar-radical-crossover cascade reaction in late stage functionalization of peptides, sugars, steroids, nucleosides and several representatives of antihistamine drugs.