Abstract Text
Introduction
Nowadays, light sheet fluorescence microscopy (LSFM) is a trendy optical imaging technique used in biology from molecular biology to tissue analysis and is highly interesting in microdevice applications. This technique possesses several advantages such as high-speed volumetric acquisition rate and low photo-toxicity that are essential for rapid 3D and 4D imaging for biological applications. Based on a simple setup design, we developed an original LSFM around a microfluidic chip and its environment to allow 3D imaging of “Organ-on-chip”-like biostructures inside microfluidic chips whatever their types or designs. Initially, this microscope was built to respond to crucial “Organ-on-Chip” issues relative to the visualization and the understanding of the vascularization occurring within the organoid-like samples. To follow up the vascularization process, we use microfluidic chips with a specific microfluidic injection system, and we reject sample preparations that would cause a loss of the sample functionality, such as clarification or fixation. The optical design promotes a large field of view at the expense of high spatial resolution, and enables to study organoids and other tissues cultured in various kind of microfluidic chips.
Set-up & Characterization
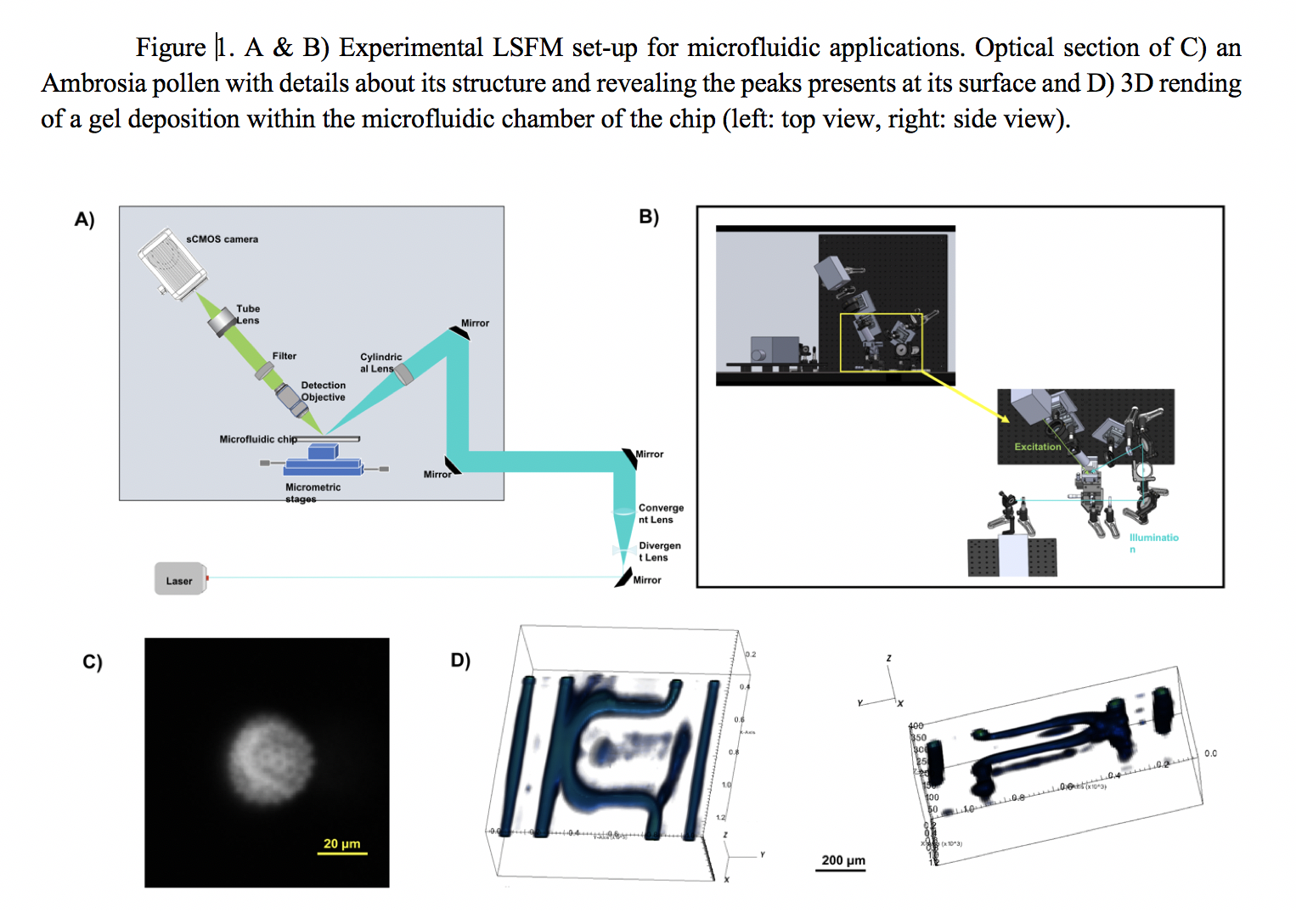
As shown in Fig.1(A&B), an expanded 488 nm Ar laser beam is shaped into a light sheet using a cylindrical lens. The propagation direction of this excitation beam creates a 45° angle with the microfluidic chip, which will necessary be maintained horizontally. The microfluidic chip can then move thanks to translation stages to scan the whole sample volume. The emitted fluorescence is collected by a long working distance 20x objective, associated with a long-pass filter and a tube lens. The wide-field image is acquired on a sCMOS camera . Illumination and detection arms are at 90° from each other. We characterized the optical performances of our system using fluorescent microspheres with a size lower than the expected resolution. Fitted Gaussian curves of the PSFs give for the lateral resolution measured at the FWHM a value of 6,5 ± 0.7μm inside the microfluidic chamber. We investigate the impact of the coverslip, the buffer solution and the induced optical aberrations on the image quality.
Results & Conclusion
To validate this system with more complex samples, we first observed diverse samples such as pollen (Fig.1.C). To demonstrate 3D imaging ability inside microfluidic chips, we imaged fluorescence gel deposits in channels of an original microfluidic design. Using a simple data stacking and a minimal data analysis, their 3D rending can then be obtained (Fig1.D). In our case, it is then possible to evaluate the thickness of the gel deposit, and then decide which working flows to use in our microfluidic experiment. We will also present the first images obtained on vascularized network.
This LSFM development leads to an innovative and relatively simple microscope dedicated to sample imaging in microfluidic chip. The system is currently under optimization to enhance the image quality and to reduce optical aberrations by modifying the incidence angles on the chips. The next step is to perform a real time 3D volume acquisition of a continuously perfused biological sample.