Abstract Text
Summary
We present an open source microscopy platform that can be configured for a wide range of microscopy modalities to deliver performance comparable to commercial research microscopes. Here we report an exemplar implementation of super-resolved HCA, demonstrating automated multiwell plate dSTORM implemented with a low-cost multimode diode lasers, a robust optical autofocus module and a new class of low-cost, cooled CMOS camera. These modules can be implemented with the open, modular openFrame microscope stand
Introduction
Fluorescence microscopy has been revolutionized by the development of advanced microscopy techniques including super-resolved imaging, quantitative phase contrast imaging, hyperspectral imaging. While SRM techniques, in particular, have transformed expectations for cell microscopy, commercial SRM instruments can be unaffordable for many researchers. We have been developing a range of self-built microscopes, including STED and dSTORM instruments and automated microscopes for high content analysis (HCA). Having worked to adapt legacy commercial microscope frames to new modalities, we realised that commercial microscope frames already present a significant cost to lower resource settings and proprietary hardware and software can add challenges to such self-build projects. Accordingly, we have developed a new, modular open source microscope frame, “openFrame”, that aims to minimise cost and to simplify the set-up of self-built advanced microscopes while providing research quality performance. Here we present an implementation of automated dSTORM microscopy with optical autofocus and open source software for image data acquisition and analysis. openFrame sits within our openScopes platform that aims to provide cost-effective access to advanced optical imaging techniques including high content analysis, super-resolved microscopy, quantitative phase contrast imaging and hyperspectral imaging.
Methods
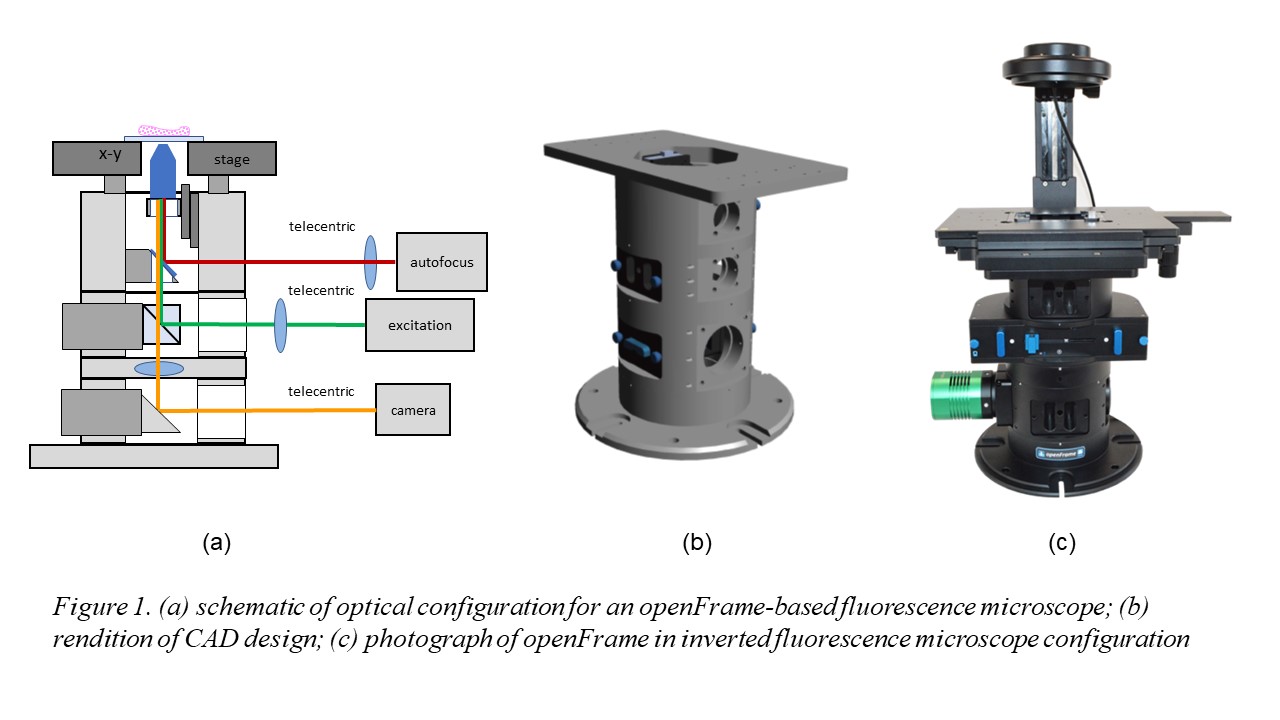
Figure 1 illustrates the openFrame concept, which is designed around a cylindrical geometry that allows straightforward centering of components along an optical axis and it is made up of different layers that can be customized as necessary for specific applications. It has been designed for straightforward manufacture using standard lathes and milling machines. openFrame has a top layer containing the objective lens mounted on a low-cost piezo-motorised stage and a beam splitter that allows an infrared laser-based autofocus unit to be deployed. We then have an excitation layer incorporating the main dichroic beamsplitter and a camera layer that includes the tube lens. This design can be easily expanded with more excitation or camera layers as required. A microscope based around the openFrame is easily maintained or modified. Furthermore, sample x-y drift measured during an acquisition of 5000 frames was less than 2 pixels (210 nm) during image acquisition, which is less than that observed with our commercial fluorescence microscope frame.
Single molecule localisation microscopy (SMLM) techniques can enable super-resolved imaging with relative simple experimental configurations based on epifluorescence or total internal reflection fluorescence (TIRF) microscopes. SMLM is particularly straightforward to implement via dSTORM [1], which is perhaps the most easily implemented technique since it can utilise common fluorophores, and several groups have demonstrated low-cost dSTORM microscopes, e.g. [2,3,4,5]. We have developed a robust and low-cost approach, easySTORM, to implement TIRF or epifluorescence SMLM microscopy on any inverted fluorescence microscope: this utilises a multimode optical fibre for efficient light coupling and the opportunity to average laser speckle (by vibrating fibre and mixing modes) for reasonably uniform illumination. The microscope can be adjusted between TIRF and epifluorescence by steering the excitation beam to be focused at different locations in the back focal plane of the objective lens. Combining this approach with low-cost, high-power multimode laser diodes to provide high power (~1 W) excitation, results in the easySTORM capability that can be added to a standard fluorescence microscope for a component cost of ~£7000 plus the cost of the objective lens and the camera. While these components can together cost £20,000 for state-of-the art components, we and others have shown that reasonable STORM images can be acquired in epifluorescence using low-cost objective lenses and with low-cost uncooled CMOS cameras. Here we demonstrated improved, cost effective performance with a fan-cooled CMOS camera.
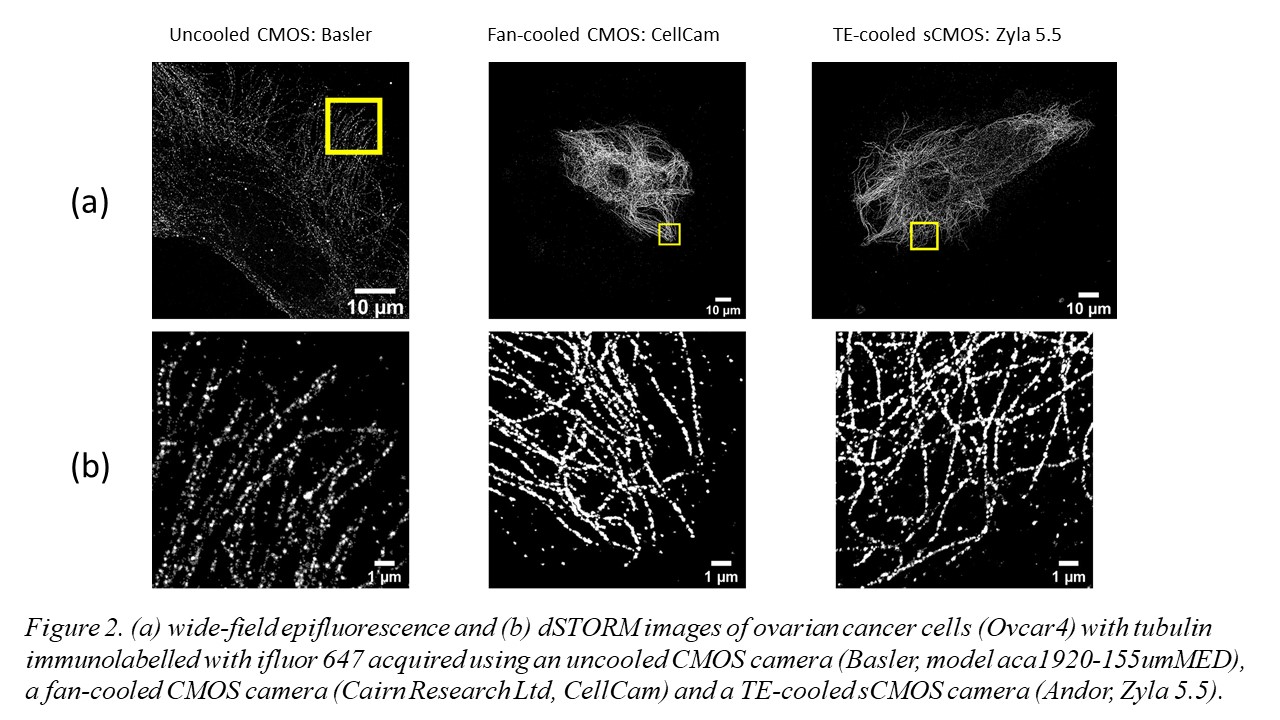
The use of multimode diode lasers improves the uniformity of illumination and provides excitation at a cost as low as £500/excitation wavelength with sufficient power to undertake STORM of samples with a field of view (FOV) >120 ×120 µm2. Such large FOV result in large (>~30 GB) data files that can require significant time (tens of minutes to hours) for SMLM data processing, e.g. using ThunderSTORM on a desktop computer. We therefore developed a parallelized SMLM analysis approach, initially based on ThunderSTORM [6] implemented on a high-performance computing cluster [7], to process SMLM data from multiple FOV in parallel on different nodes in the HPC cluster or to accelerate the processing of SMLM data from one FOV by dividing the localisation task between multiple nodes. The ability of easySTORM to image large FOV combined with the ability to scale up the SMLM data processing rate using HPC resources are key enablers for high throughput SMLM, noting this has previously been demonstrated with PALM [8] and STORM [9]. We have developed a low-cost open source automated SMLM high content analysis platform combining easySTORM with an optical autofocus and motorised stage-scanning to enable automated multiwell plate dSTORM acquisition [10]. The autofocus module utilises a convolutional neural network (CNN) that can robustly determine the distance from focus by analysing a single image captured on the autofocus camera . Automated dSTORM has been applied to high content super-resolved imaging, including of focal adhesions in melanoma cells, and phagocytosis of bacteria. We have also explored the use of a new generation of fan-cooled CMOS cameras for SMLM, noting that the relatively high frame rate used for SMLM means that fan-cooling can provide similar SMLM performance to thermoelectric cooled sCMOS cameras, as illustrated in figure 2.
Conclusions
We have presented an open microscopy platform applicable to most imaging modalities, including automated and super-resolved microscopy. Links to the open-source software to control the easySTORM microscope and for the scripts for the HPC processing of SMLM data will be provided at: https://www.imperial.ac.uk/photonics/research/biophotonics/instruments--software/open-source-software/. Further information can be found at www.openScopes.com.
Keywords
open source, fluorescence microscopy
References
[1] Heilemann, M., et al, Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angewandte Chemie (International Ed. in English), 47, 6172–6176 (2008)
[2] Holm, T., et al. “A blueprint for cost-efficient localization microscopy,” Chem-PhysChem. 15, 651–654 (2014).
[3] Kwakwa, K., et al.,. “easySTORM: a robust, lower-cost approach to localisation and TIRF microscopy,”J. Biophotonics, 9, 948–957 (2016).
[4] Ma, H., Fu, R., Xu, J., & Liu, Y. A simple and cost-effective setup for super-resolution localization microscopy. Scientific Reports, 7, 1542 (2017).
[5] Diekmann, R., et al., “Characterization of an industry-grade CMOS camera well suited for single molecule localization microscopy - High performance super-resolution at low cost,” Scientific Reports, 7, 14425 (2017).
[6] Ovesný, et al., “ThunderSTORM: A comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging,” Bioinformatics, 30, 2389–2390 (2014).
[7] Munro, I., et al., “Accelerating single molecule localization microscopy through parallel processing on a high‐performance computing cluster,” J. Microscopy 273 148-160 (2019)
[8] Holden, et al., S. “High throughput 3D super-resolution microscopy reveals Caulobacter crescentus in vivo Z-ring organization,” Proc. Natl. Acad. Sci. U.S.A. 111, 4566–4571 (2014).
[9] Beghin, A., et al. “Localization-based super-resolution imaging meets high-content screening,”. Nature Methods, 14, 1184–1190 (2017).
[10] J. Lightley et al, “Robust optical autofocus system utilizing neural networks trained for extended range and time-course and automated multiwell plate imaging including single molecule localization microscopy”, bioRxiv (2021), https://doi.org/10.1101/2021.03.05.431171