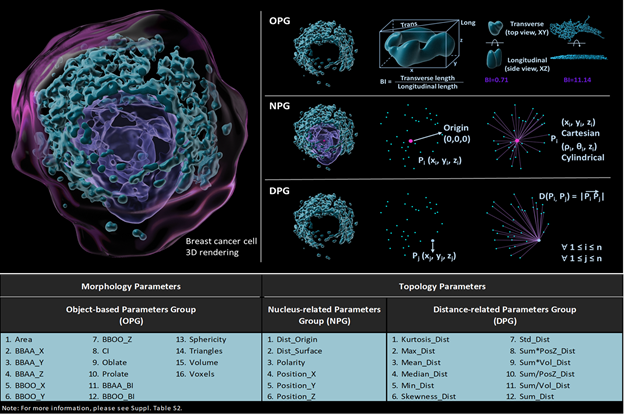
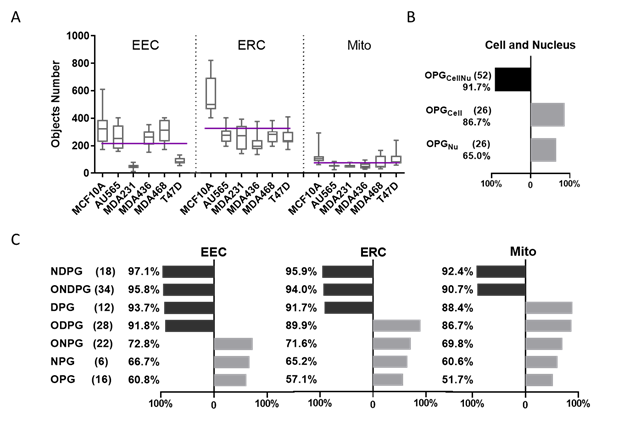
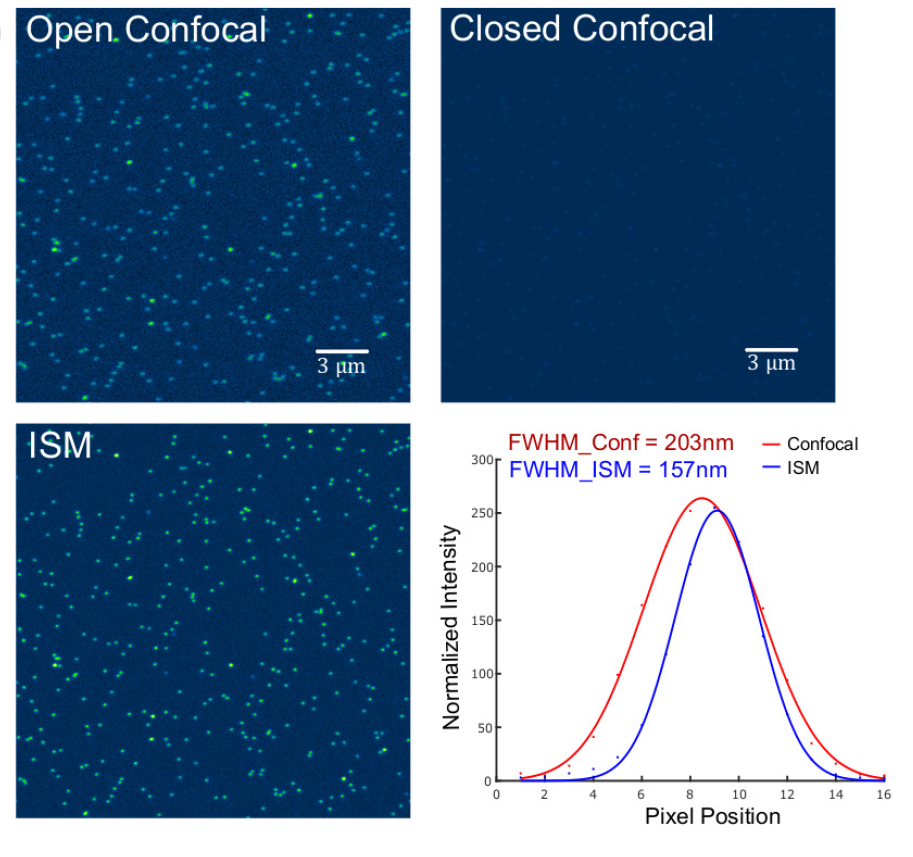
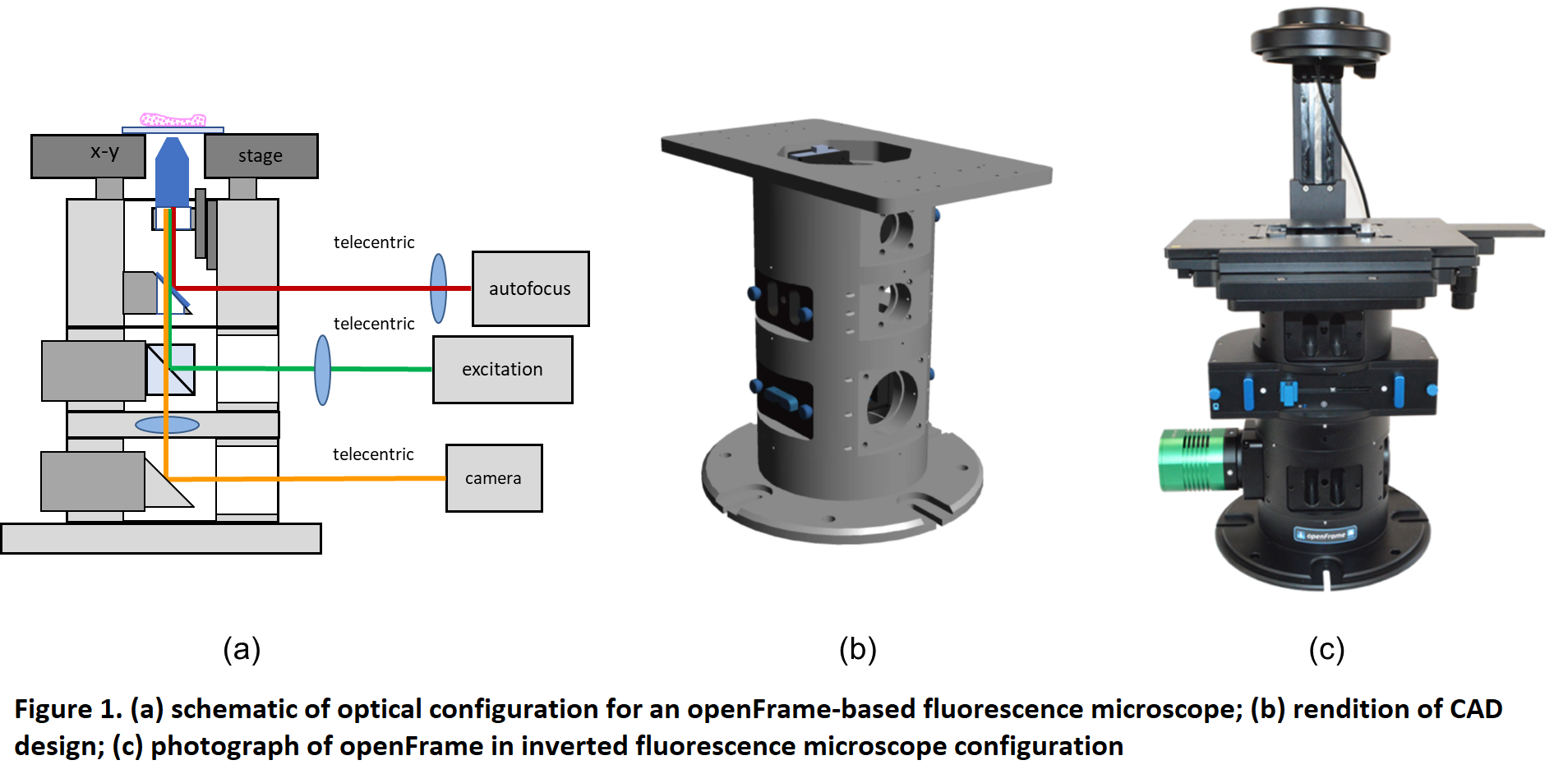
Abstract Text
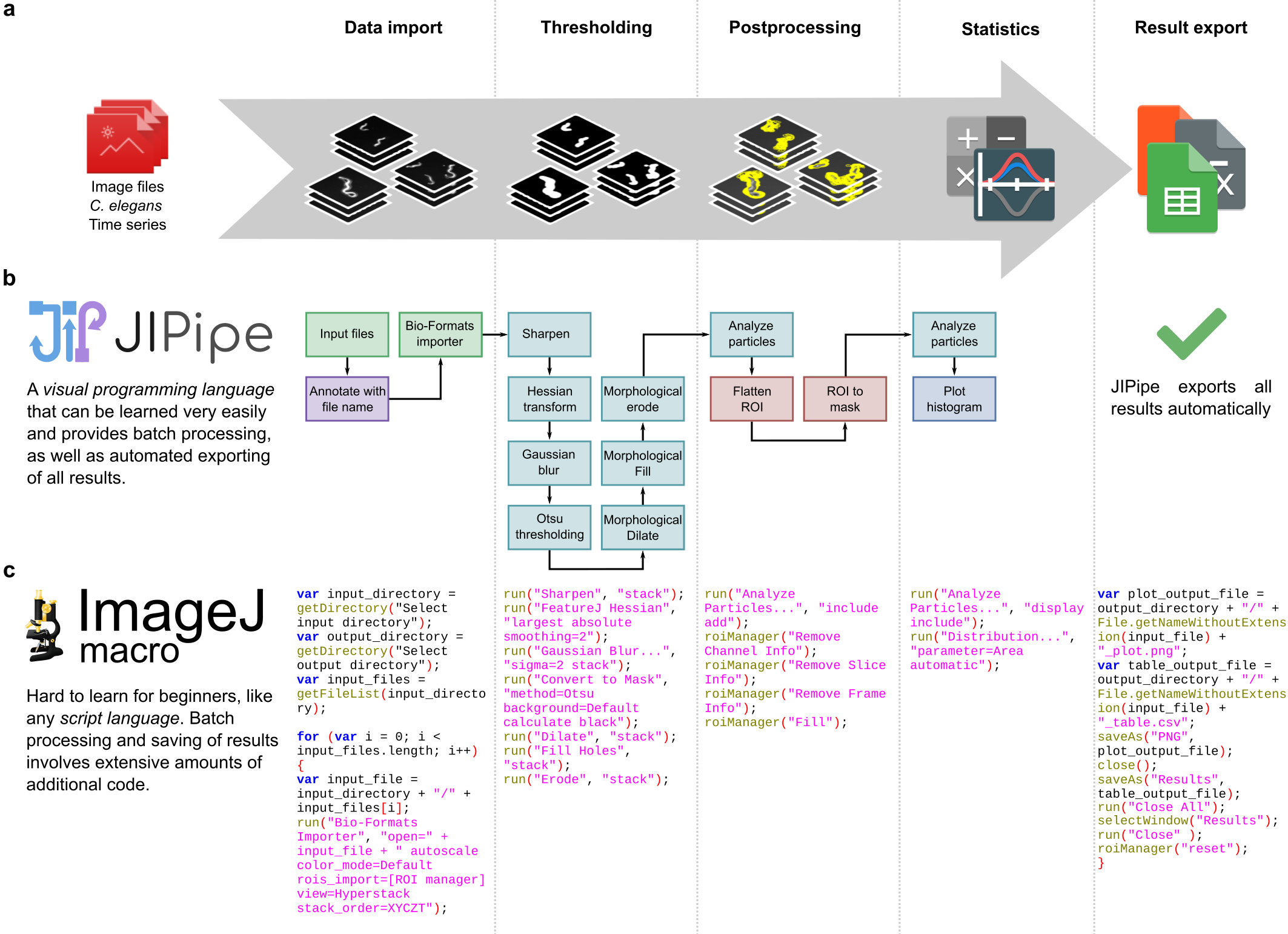
The goal of our study is to develop label-free method for measuring the organization of amyloid fibrils based on their polarization-resolved two-photon excited autofluorescence. For our purpose, we have modified the model used for measuring fibril organization from polarization resolved one-photon fluorescence microscopy and applied it to successfully determine the internal structural organization of fibrils inside insulin spherulites.
Amyloids are ordered protein aggregates and a hallmark of multiple neurodegenerative disorders such as Parkinson’s or Alzheimer’s. Understanding the origin and course of amyloidogenesis is a key issue for creation of effective curative methods. A plethora of information about this process could be gathered from studying the organization of amyloid structures in different conditions.[1] In previous years, it was shown that ordering of amyloids stained with Thioflavin T could be efficiently determined using polarization-resolved fluorescence microscopy. [2] However, a label influences the aggregation process [3] and the data obtained from stained structures may differ from the naturally occurring label-free ones. Moreover, correct determination of fibril ordering in densely packed superstructures like amyloid plaques needs high precision which could hardly be achieved using standard fluorescence microscopes. Our method is overcoming this drawbacks, allowing to determine the local ordering of label-free amyloid fibrils in spherulites, amyloid superstructures, using their intrinsic polarization-resolved two-photon excited autofluorescence. [4] However, a new model was needed to analyse the obtained data. We have adapted the theory used with polarization-resolved fluorescence microscopy to two-photon microscopy and successfully applied it for calculating the arrangement of fibrils and their fluorophores in densely packed amyloid spherulites.
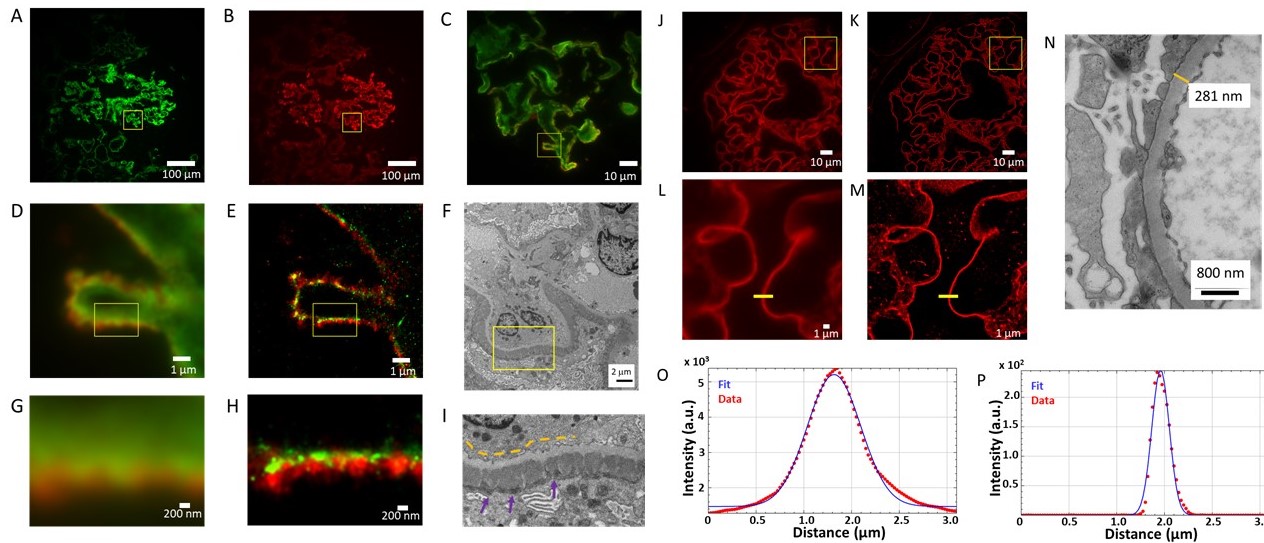
The sample preparation along with measurement setup and procedure is described in Supporting Information of our aforementioned paper. [4] To calculate the ordering of a spherulite structure we have successfully implemented theoretical models previously used by our group to calculate organization of DNA strands [5] which is a modification of model used for polarization-resolved fluorescence microscopy. [2] Data analysis was performed in Spyder Python 3 IDE. Fitting the theoretical models into experimental data was done using correlation of various model parameters such as shape, intensity, angle maxima location and checked for maximal R2 coefficient. Functions were based on NumPy and SciPy Python libraries.
The model assumes that total intensity of two-photon excited fluorescence is dependent on the probability of emission and two-photon absorption, intertwined with molecular distribution function, which is dependent on three structural parameters: rotation of the fibril in XY sample plane, orientational distribution of the emission dipole with respect to the long fibril axis and emission dipole orientational distribution aberrations due to the molecular rotations in filaments (Fig.1). Thus, polarization resolved two-photon fluorescence from highly ordered spots on sample where the average fibril orientation is known could be used to calculate the orientational distribution of fibrils. Appling this model to insulin spherulites have already shown differences between the calculated emission dipole orientational distribution of label-free and stained aggregates or that increasing the salt concentration narrows the emission dipole moment orientational distribution, which was in agreement with calculations of single fibrils imaged using Atomic Force Microscope.
In conclusion, we have successfully modified and implemented the model used for polarization resolved fluorescence microscopy to determine the orientational distribution of autofluorescence in amyloids, measured under two-photon microscopy and showed that it differs depending on salt concentration or presence of a dye. Moreover, the presence of polarization dependent alignment of autofluorescence dipole moments supports the mechanism of amyloid autofluorescence, proposed by Grisanti et al. [6] moving us one step closer to understand the origin of amyloid autofluorescence process.
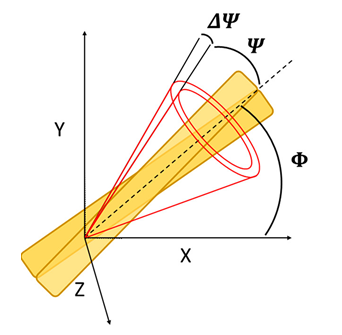
Figure 1. Open cone model of the conical distribution of the emission dipole of the dye (half angle, Ψ) in respect to the long fibril axis (dashed line). Rotation of the fibril in the XY microscope sample plane is described by the Φ angle. Aberrations of Ψ due to the molecular rotations in filaments are described by ΔΨ.
References
[1] M. G. Iadanza, M. P. Jackson, E. W. Hewitt, N. A. Ranson, and S. E. Radford, “A new era for understanding amyloid structures and disease,” Nat. Rev. Mol. Cell Biol., vol. 19, no. 12, pp. 755–773, 2018
[2] J. Duboisset, P. Ferrand, W. He, X. Wang, H. Rigneault, and S. Brasselet, “Thioflavine-T and Congo Red Reveal the Polymorphism of Insulin Amyloid Fibrils When Probed by Polarization-Resolved Fluorescence Microscopy,” J. Phys. Chem. B, vol. 117, no. 3, pp. 784–788, Jan. 2013
[3] M. I. Sulatsky, A. I. Sulatskaya, O. I. Povarova, I. A. Antifeeva, I. M. Kuznetsova, and K. K. Turoverov, “Effect of the fluorescent probes ThT and ANS on the mature amyloid fibrils,” Prion, vol. 14, no. 1, pp. 67–75, Dec. 2020
[4] P. Obstarczyk, M. Lipok, M. Grelich-Mucha, M. Samoć, and J. Olesiak-Bańska, “Two-Photon Excited Polarization-Dependent Autofluorescence of Amyloids as a Label-Free Method of Fibril Organization Imaging,” J. Phys. Chem. Lett., vol. 12, no. 5, pp. 1432–1437, Feb. 2021
[5] H. Mojzisova, J. Olesiak, M. Zielinski, K. Matczyszyn, D. Chauvat, and J. Zyss, “Polarization-Sensitive Two-Photon Microscopy Study of the Organization of Liquid-Crystalline DNA,” Biophys. J., vol. 97, no. 8, pp. 2348–2357, 2009
[6] L. Grisanti, M. Sapunar, A. Hassanali, and N. Došlić, “Toward Understanding Optical Properties of Amyloids: A Reaction Path and Nonadiabatic Dynamics Study,” J. Am. Chem. Soc., vol. 142, no. 42, pp. 18042–18049, Oct. 2020