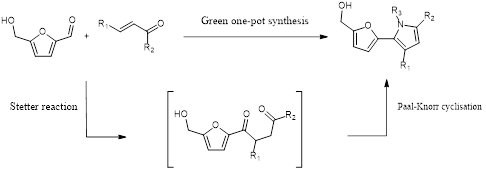
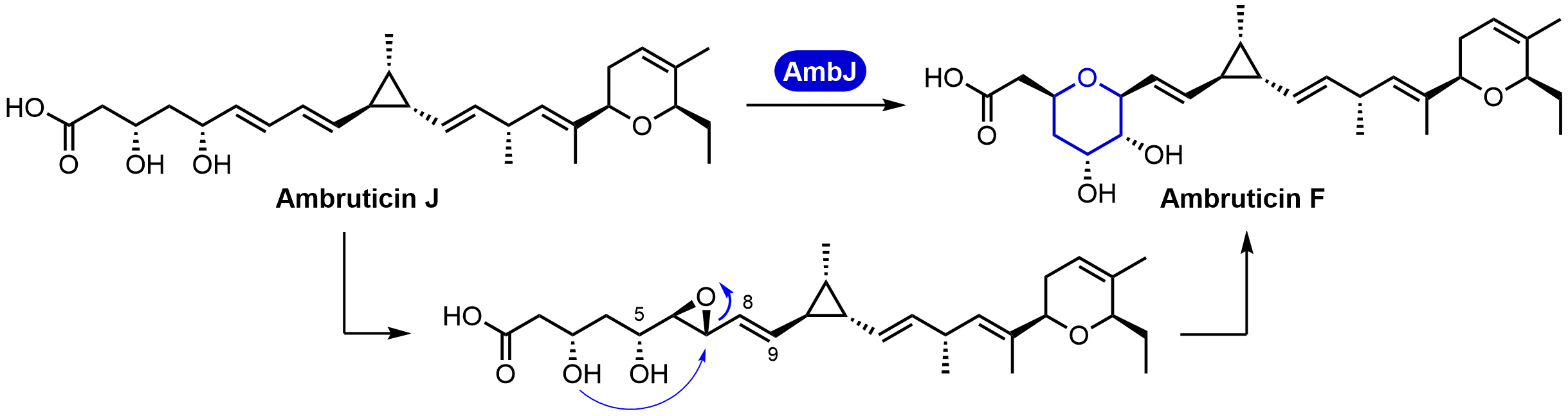
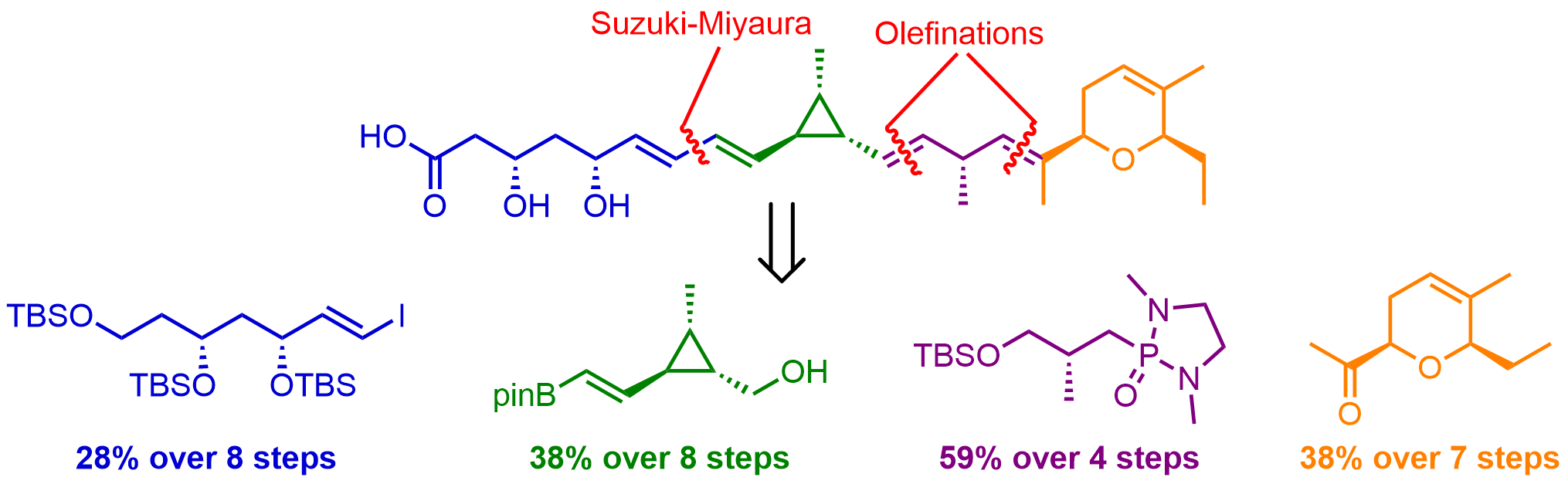
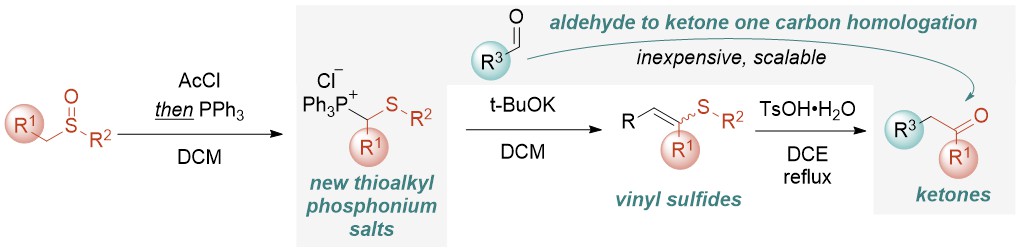
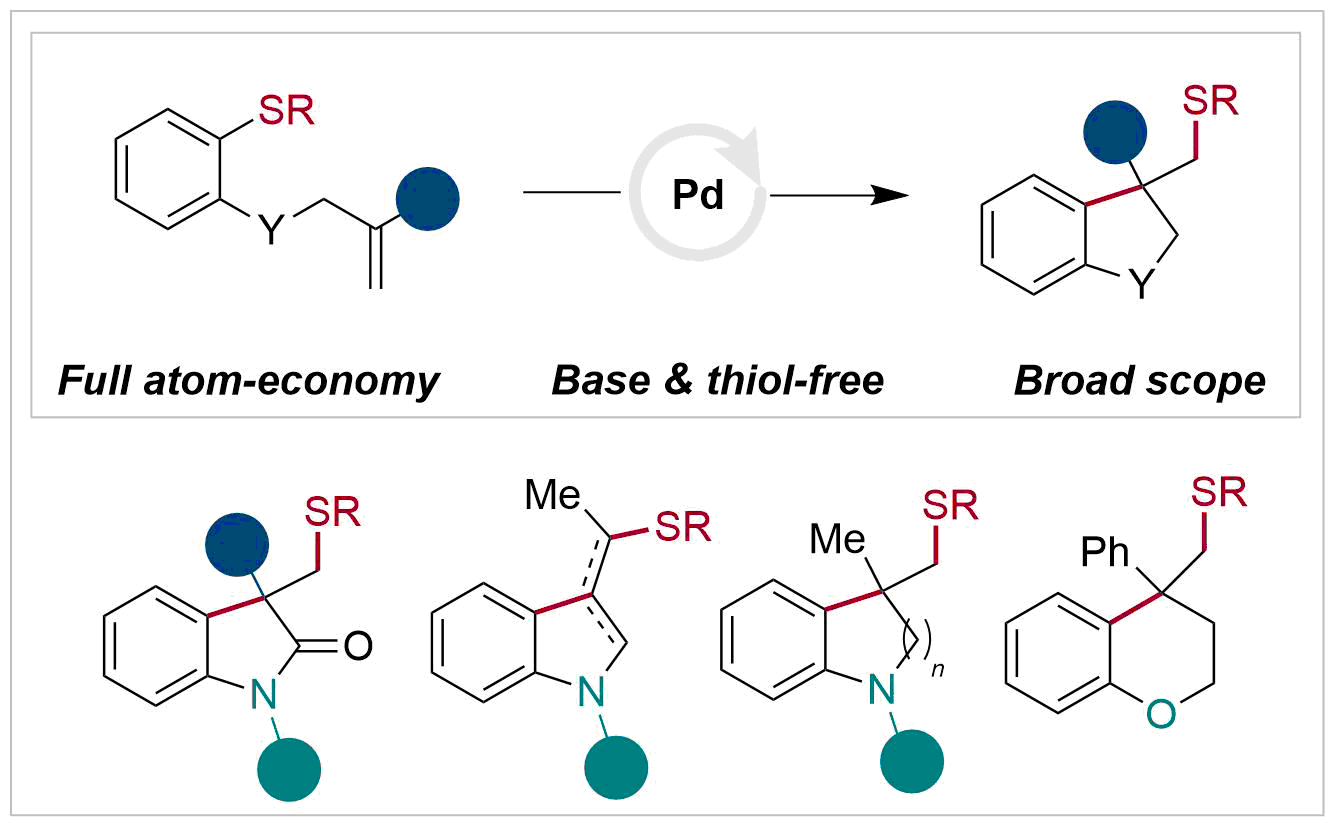
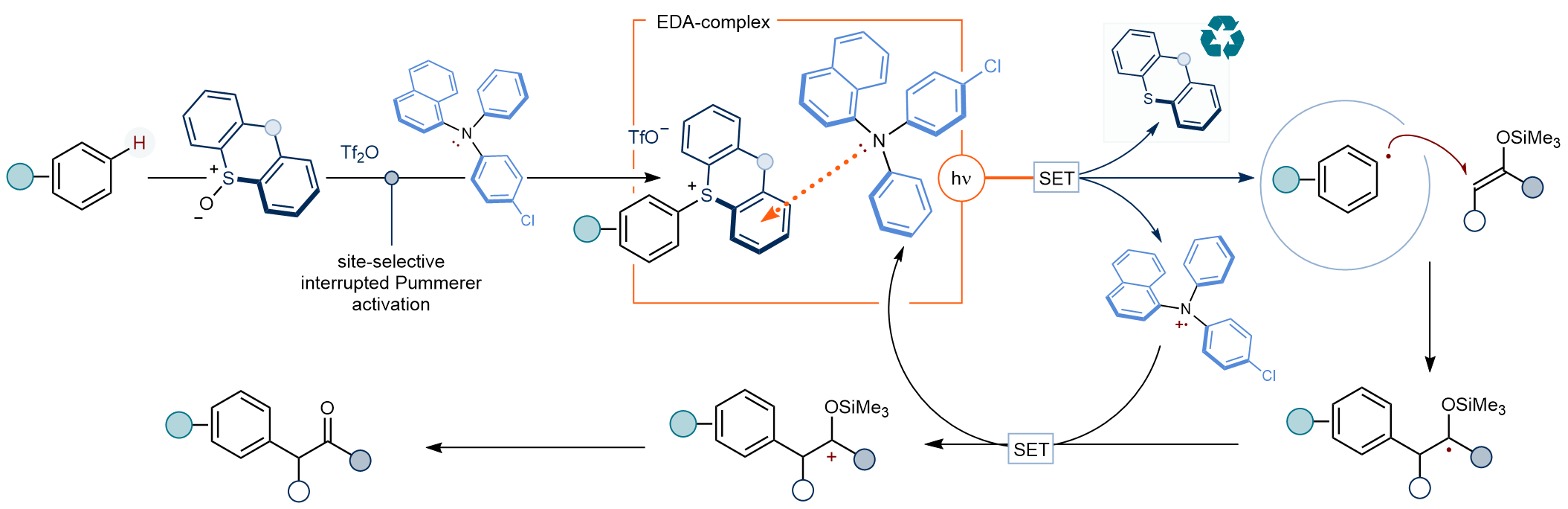
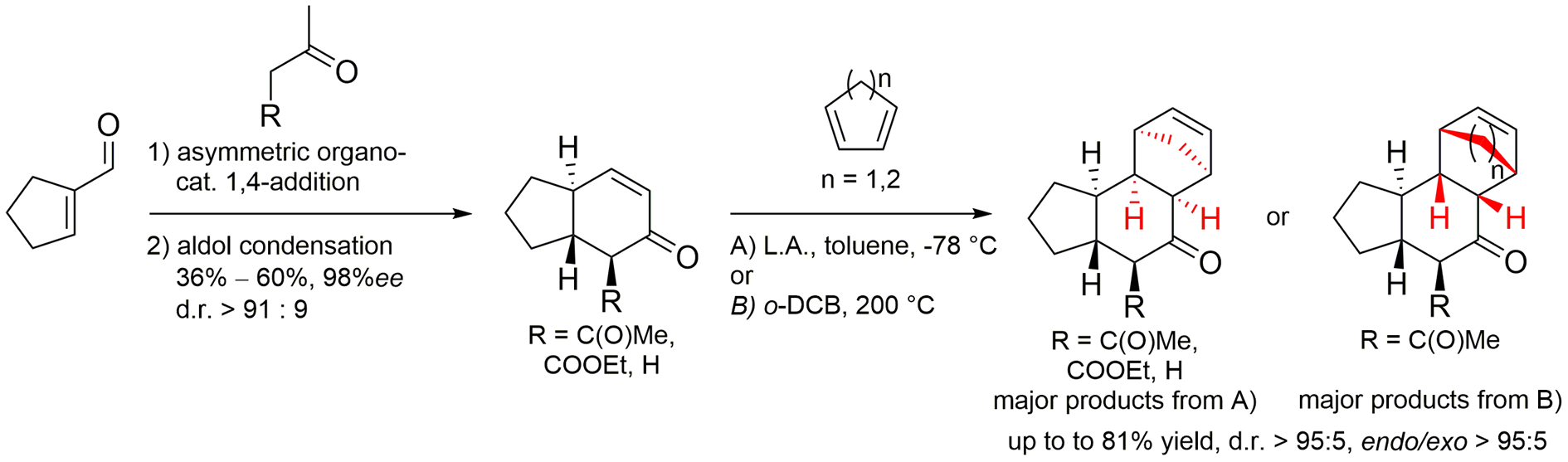
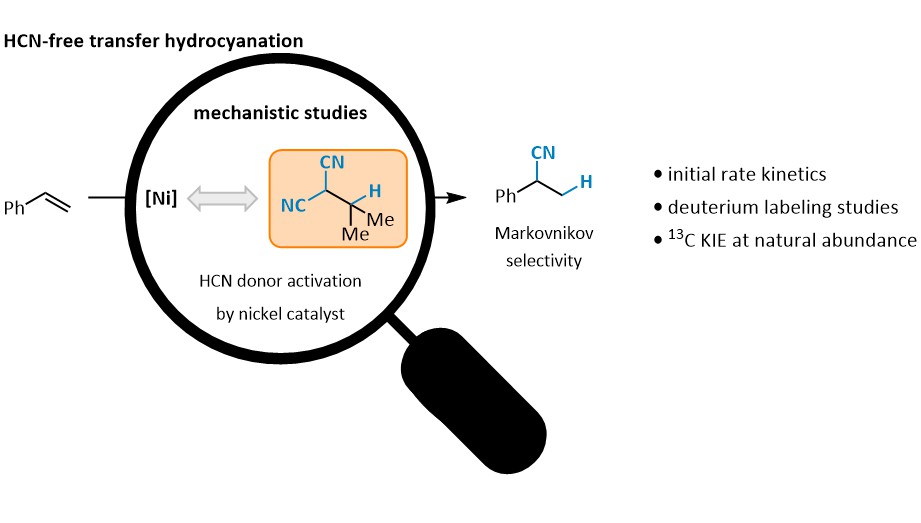
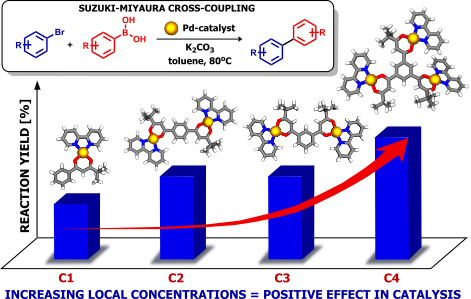
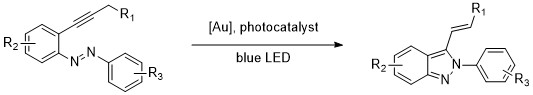
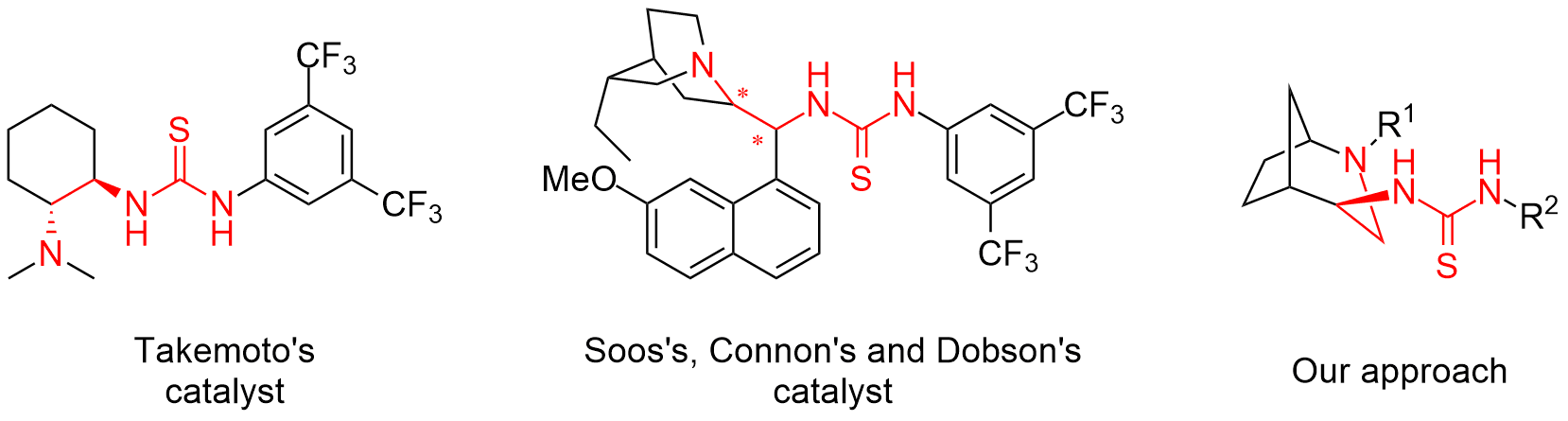
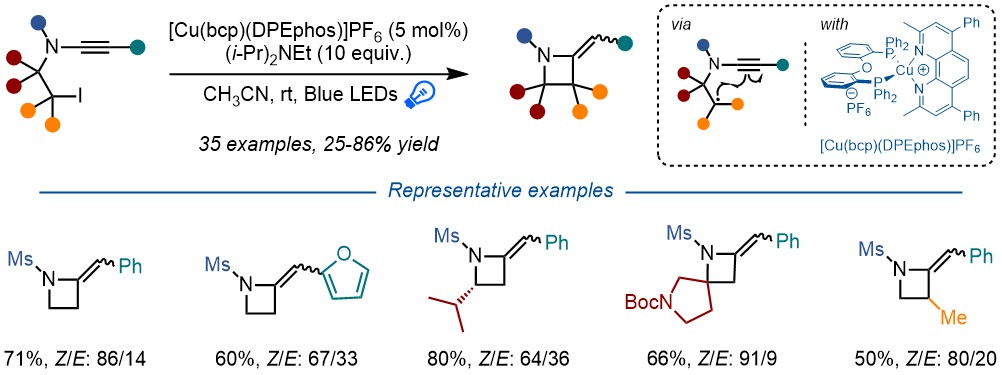
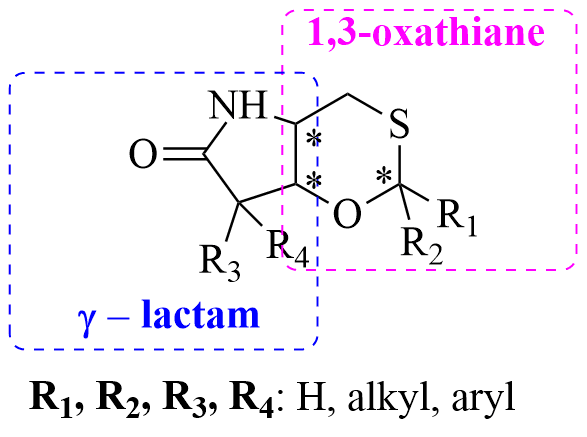
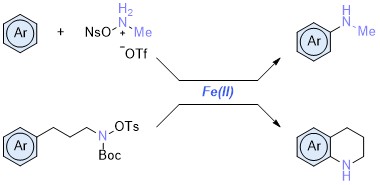
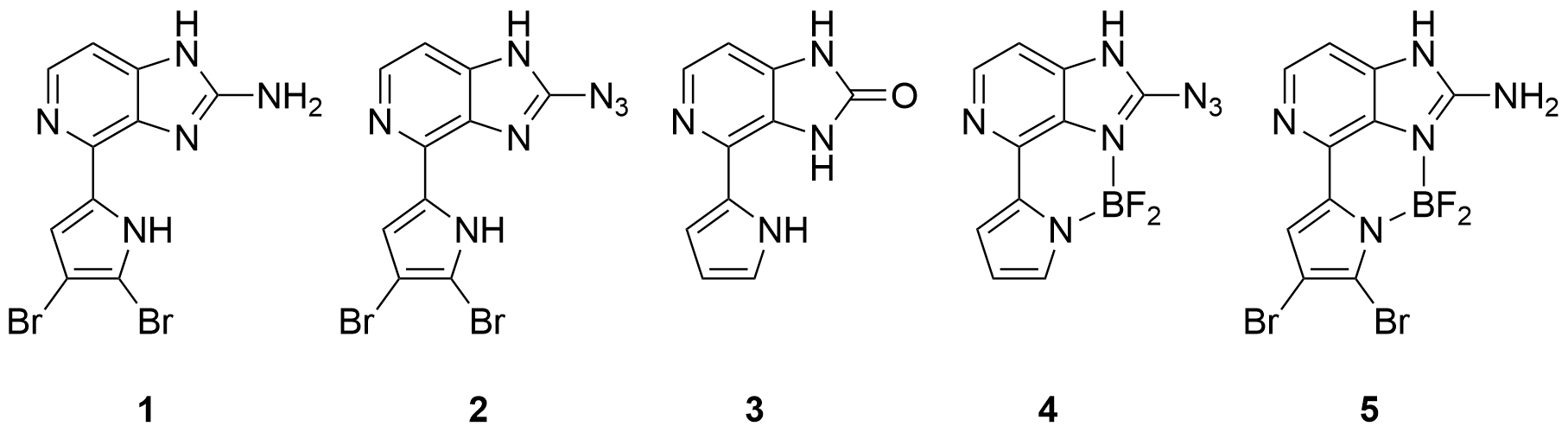
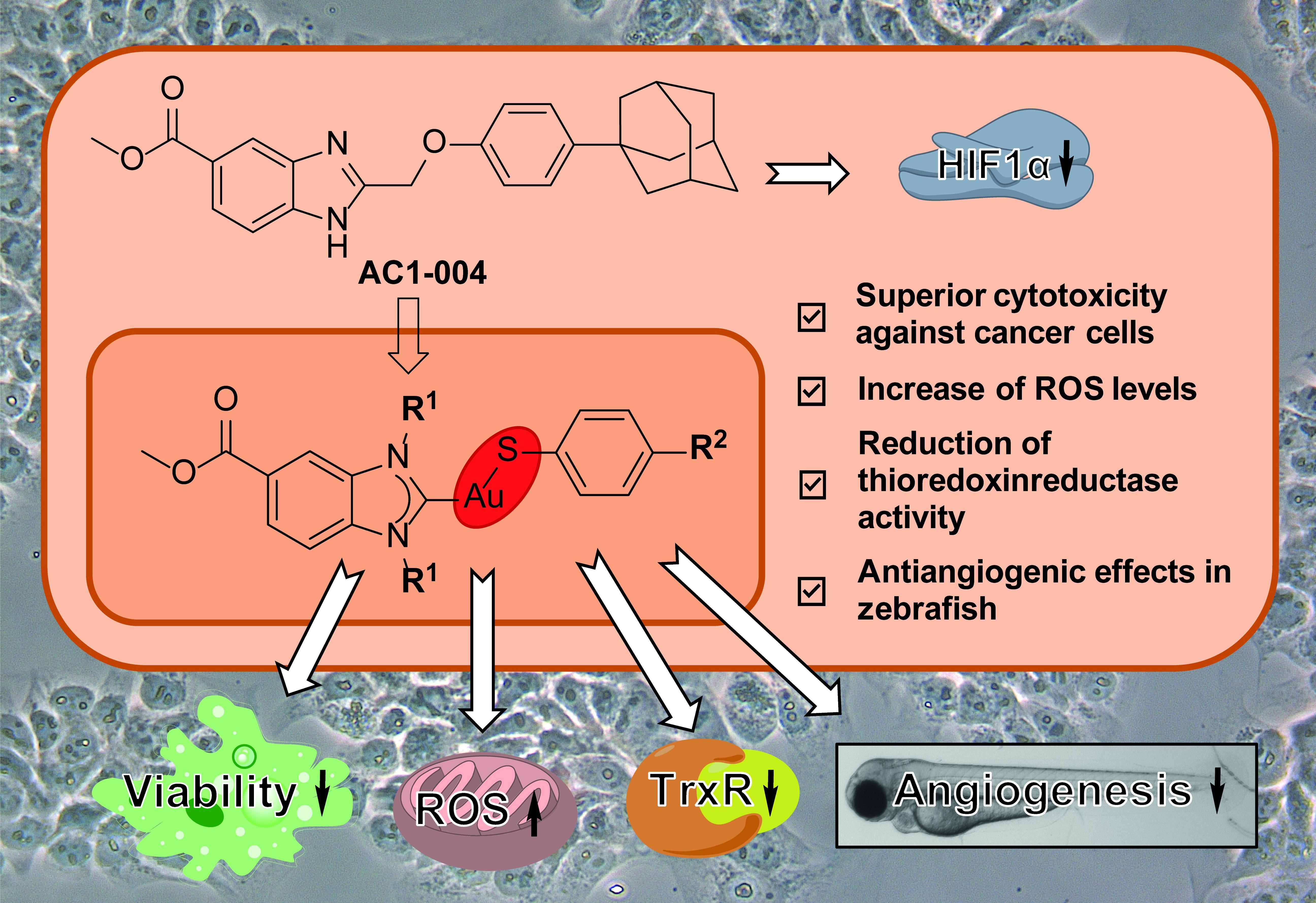
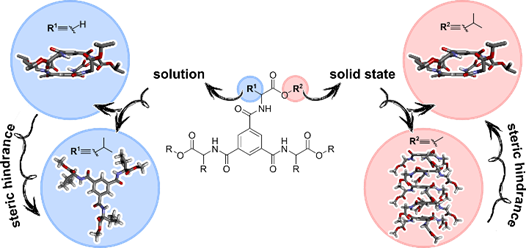
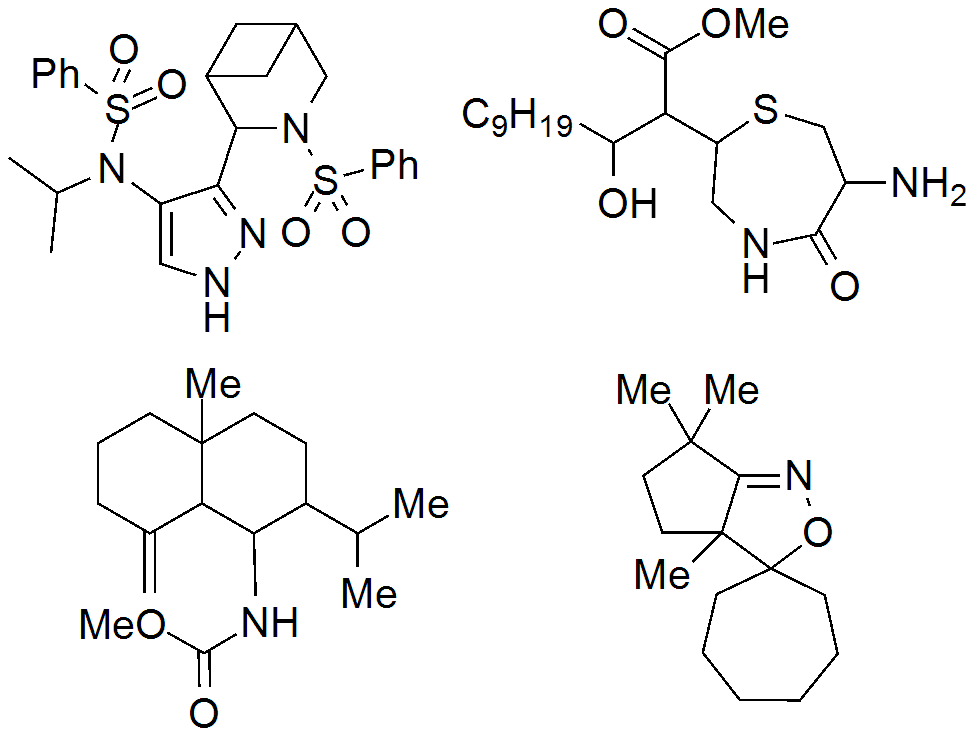
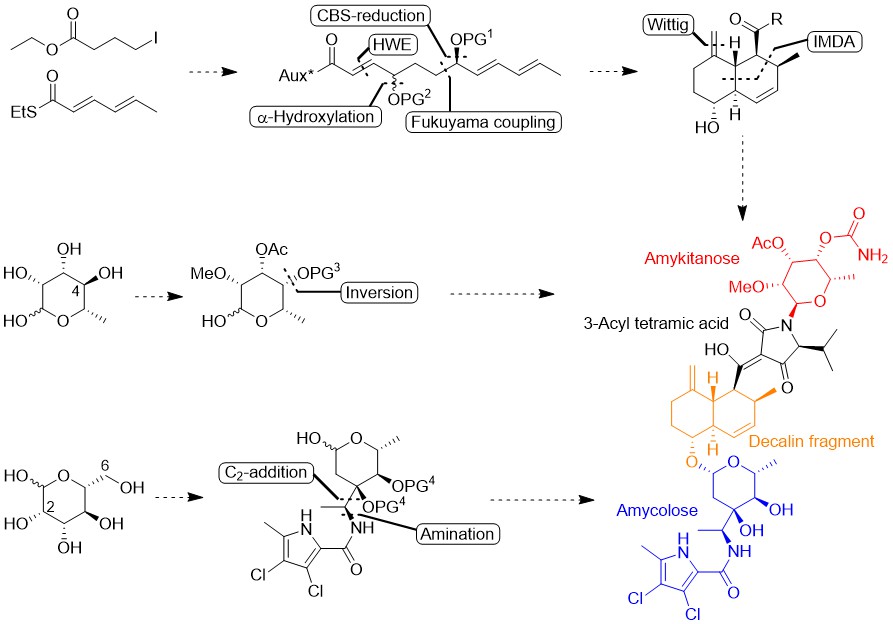
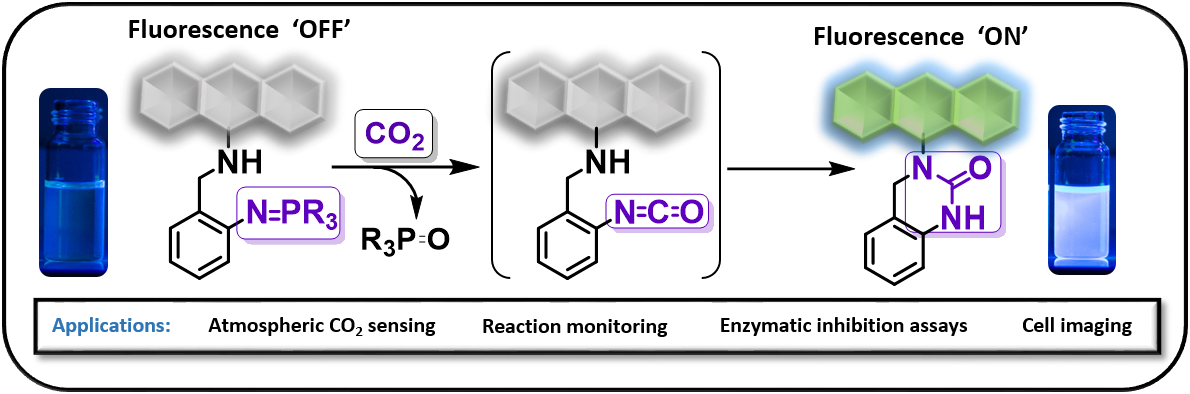
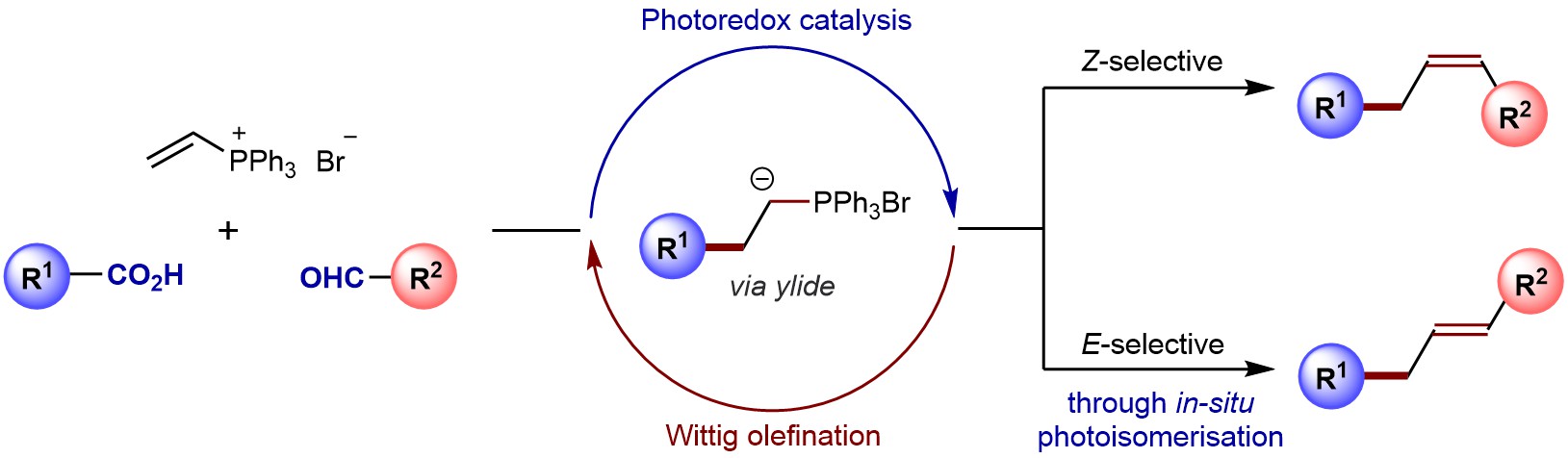
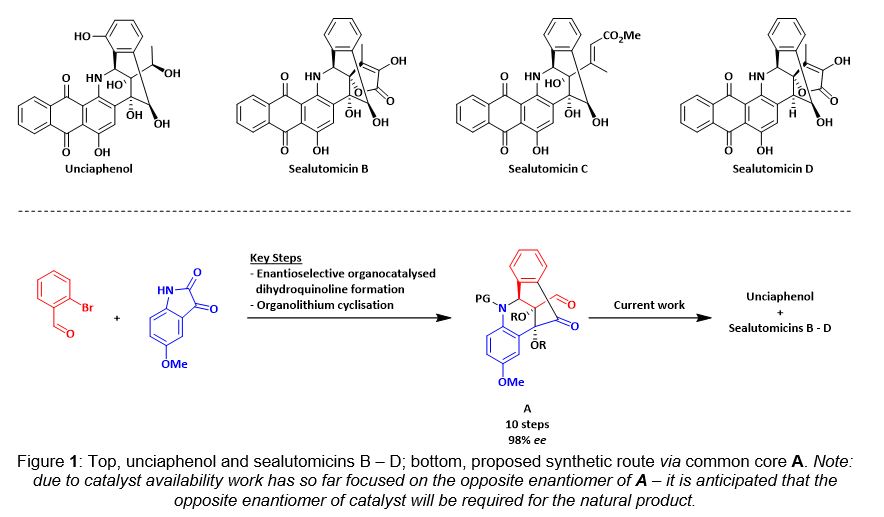
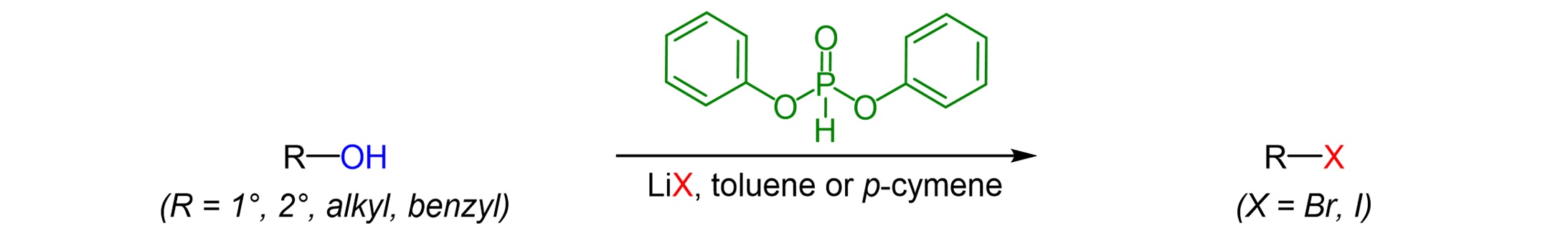
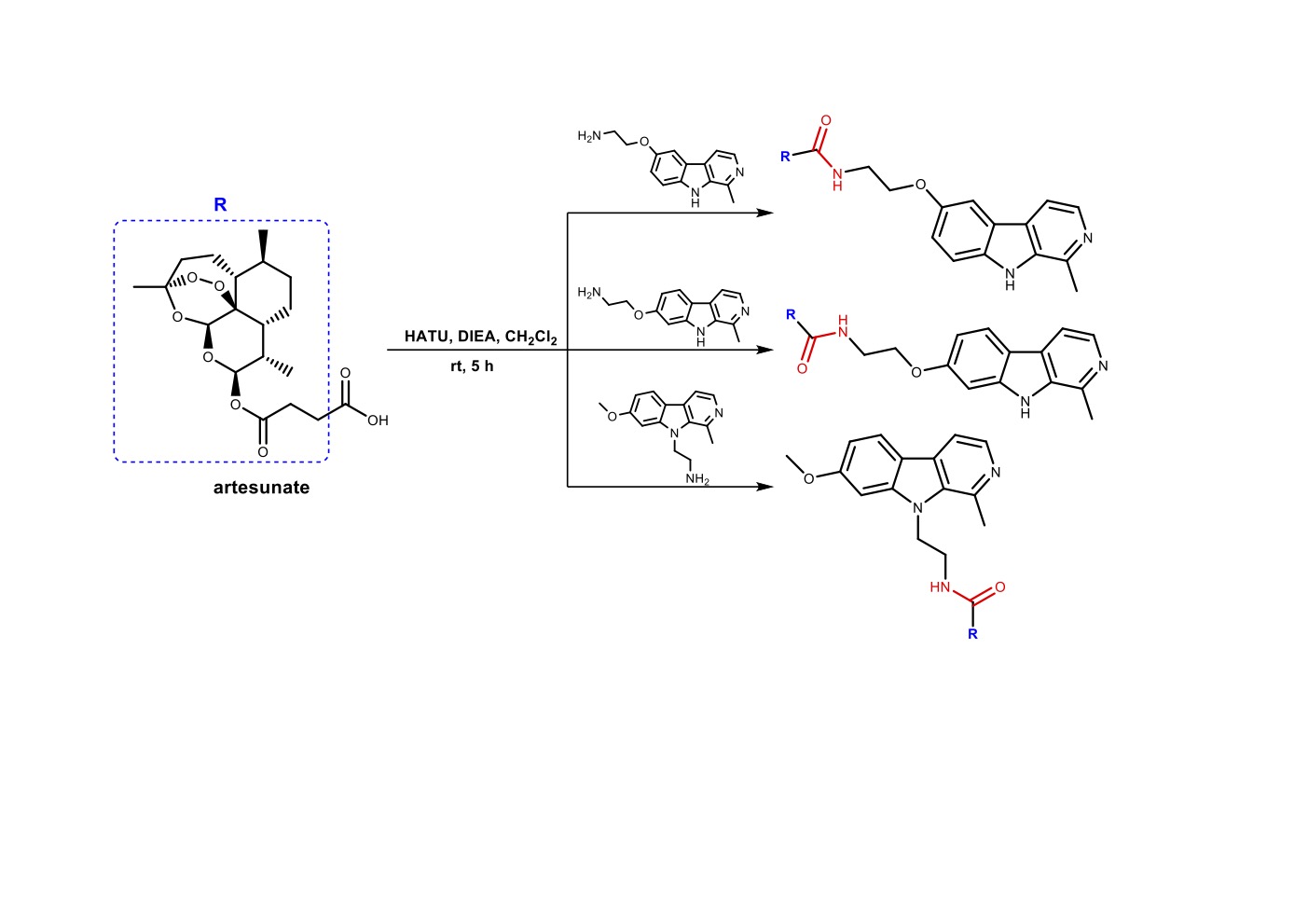
Abstract
The activation and functionalisation of carbon–fluorine bonds represents a significant synthetic challenge, given the high thermodynamic barrier to C–F bond cleavage. Stoichiometric hydridoborane-mediated C–F functionalisation has recently emerged, but is yet to be rendered catalytic.[1–3] This talk will discuss the borane-catalysed coupling of alkyl fluorides with arenes (carbon–carbon bond formation) and carboxylic acids (carbon–oxygen bond formation) using transborylation reactions to achieve catalytic turnover.[4] Successful C–C and C–O coupling across a variety of structurally and electronically differentiated arenes and carboxylic acids was achieved using 9-borabicyclo[3.3.1]nonane (H-B-9-BBN) as the catalyst and pinacolborane (HBpin), with excellent functional group tolerance, and was highly applicable to late-stage esterification of active pharmaceutical ingredients. Experimental and computational studies suggest a mechanistic dichotomy for the carbon–carbon and carbon–oxygen coupling reactions. B–F transborylation (B–F/B–H metathesis) between F-B-9-BBN and HBpin enabled catalytic turnover for carbon–carbon bond formation, whereas direct exchange between the alkyl fluoride and acyloxyboronic ester (C–F/B–O metathesis) was proposed for carbon–oxygen coupling, where H-B-9-BBN catalysed the dehydrocoupling of the carboxylic acid with HBpin, representing a novel C‒F activation pathway for organoboranes.
References
[1] K.L. Bamford, S.S. Chitnis, Z. Qu, D.W. Stephan, Chem. – A Eur. J. 24 (2018) 16014–16018.
[2] J. Guo, K.L. Bamford, D.W. Stephan, Org. Biomol. Chem. 17 (2019) 5258–5261.
[3] N.A. Phillips, J. O’Hanlon, T.N. Hooper, A.J.P. White, Org. Lett. 21 (2019) 7289–7293.
[4] D.R. Willcox, G.S. Nichol, S.P. Thomas, ACS Catal. 11 (2021) 3190–3197.
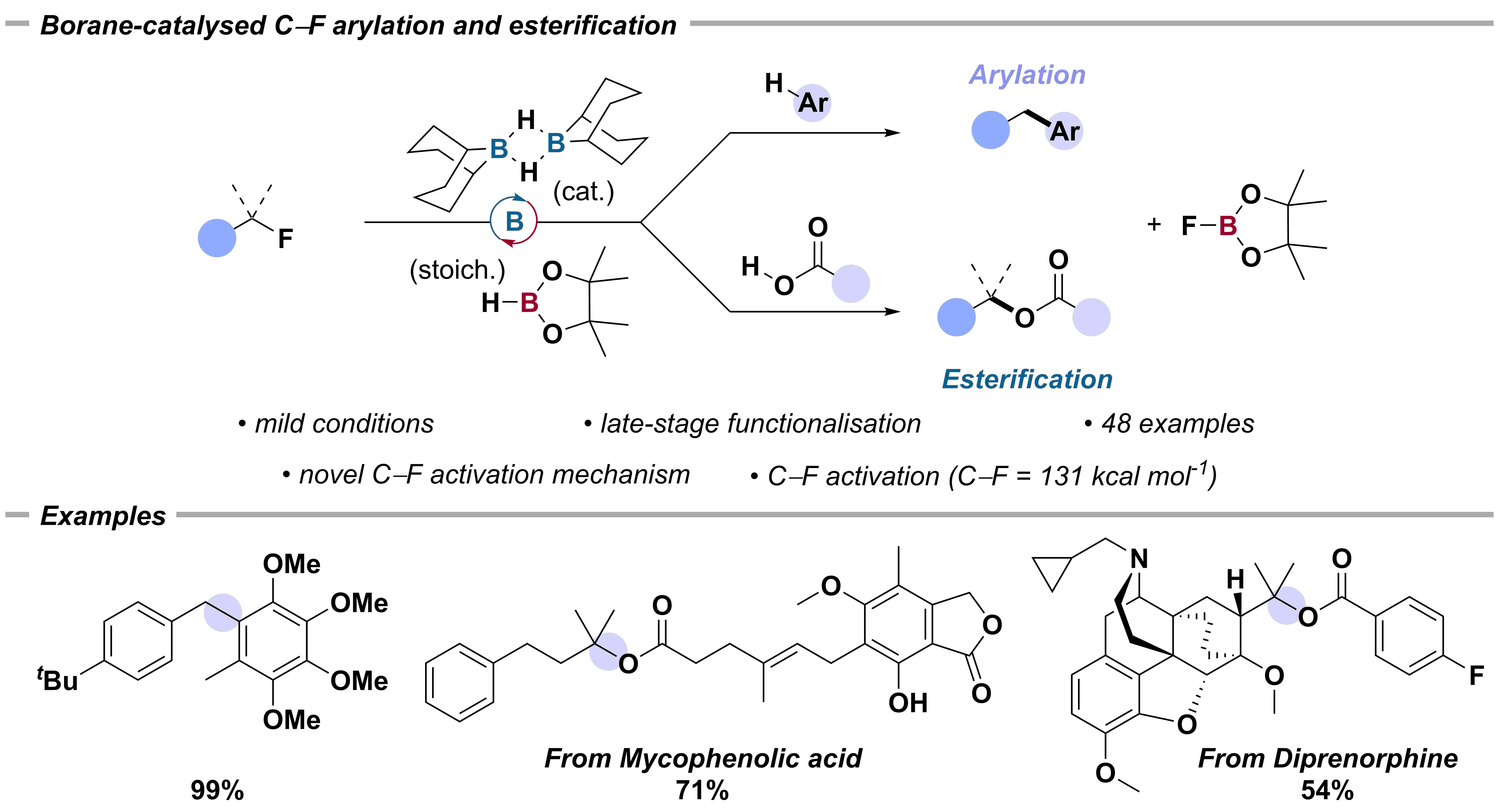
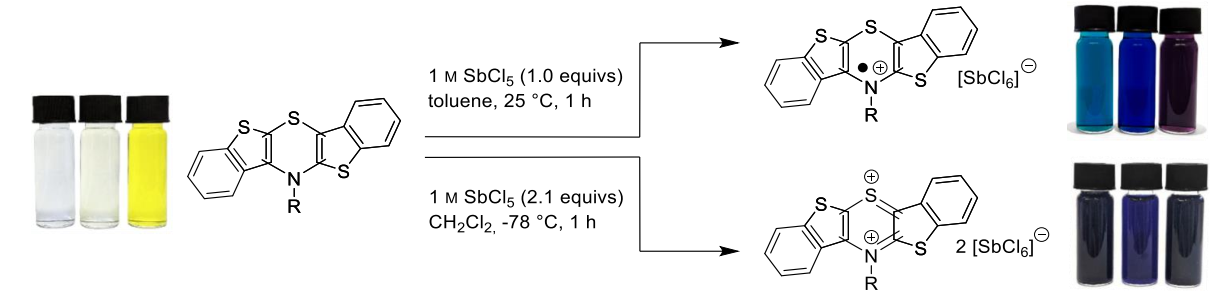 Figure 1: Chemical oxidation of syn-syn, syn-anti and anti-anti regioisomer of N-p-Fluorophenyl-BBTT.
Figure 1: Chemical oxidation of syn-syn, syn-anti and anti-anti regioisomer of N-p-Fluorophenyl-BBTT.