Abstract Text
We study lightguiding in nanowires and protein-based molecular motors using fluorescence microscopy. Due to optical wavelengths resonance, III-V GaP nanowires emit fluorescence from fluorophores located in proximity of their surface (Fig. 1A), which we have employed for molecular sensing [1]. We demonstrate that lightguiding depends on nanowire dimensions and fluorescence wavelength. Also, our modelling shows up to 50x increase in emission for fluorophores on nanowire surface, which we expect to be accompanied by stronger bleaching. Initial experiments show that, upon the increase of excitation power, the bleaching rates of Alexa Fluor 647 increase differently for nanowires with diameter in range 50 – 250 nm.
As an application of lightguiding in nanowires, we aim to demonstrate stepping of an artificial protein-based molecular motor, the Tumbleweed [2]. The motor is built of protein domains which bind to sequences on a DNA oligonucleotide track in presence of S-Adenosyl methionine, tryptophane, and NiCl2. Tumbleweed steps over the distance of about 10 nm upon the change of ligand. If nanowires are decorated with short DNA tracks, fluorescently labelled molecular motors can be detected when moving towards a nanowire surface, as when motors are close enough to the surface, lightguiding can occur (Fig. 1B).
We are also working on resolving the motor steps, which requires sub-diffraction resolution. Inspired with the concept of surface optical profilometry [3], we attach multiple DNA tracks to micron-sized beads to localize the position of fluorescently labelled motors as a centre of fluorescence pattern around the beads (Fig. 1C). We demonstrate the specificity of binding, and that the motors remain bound to tracks upon the change of ligands.
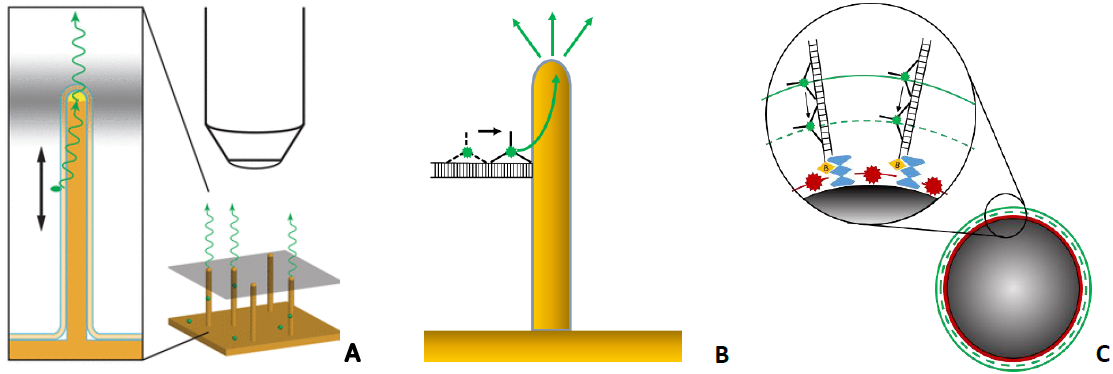
Figure 1. A – lightguiding in a nanowire; B – molecular motor on DNA track attached to a nanowire; C –molecular motors on DNA tracks on a bead.
References
[1] D. Verardo et al., “Nanowires for Biosensing: Lightguiding of Fluorescence as a Function of Diameter and Wavelength,” Nano Lett., vol. 18, no. 8, pp. 4796–4802, 2018, doi: 10.1021/acs.nanolett.8b01360.
[2] E. H. C. Bromley et al., “The tumbleweed: Towards a synthetic protein motor,” HFSP J., vol. 3, no. 3, pp. 204–212, Jun. 2009, doi: 10.2976/1.3111282.
[3] M. H. Bakalar, A. M. Joffe, E. M. Schmid, S. Son, M. Podolski, and D. A. Fletcher, “Size-Dependent Segregation Controls Macrophage Phagocytosis of Antibody-Opsonized Targets,” Cell, vol. 174, no. 1, pp. 131-142.e13, 2018, doi: 10.1016/j.cell.2018.05.059.