Photo upload

Abstract
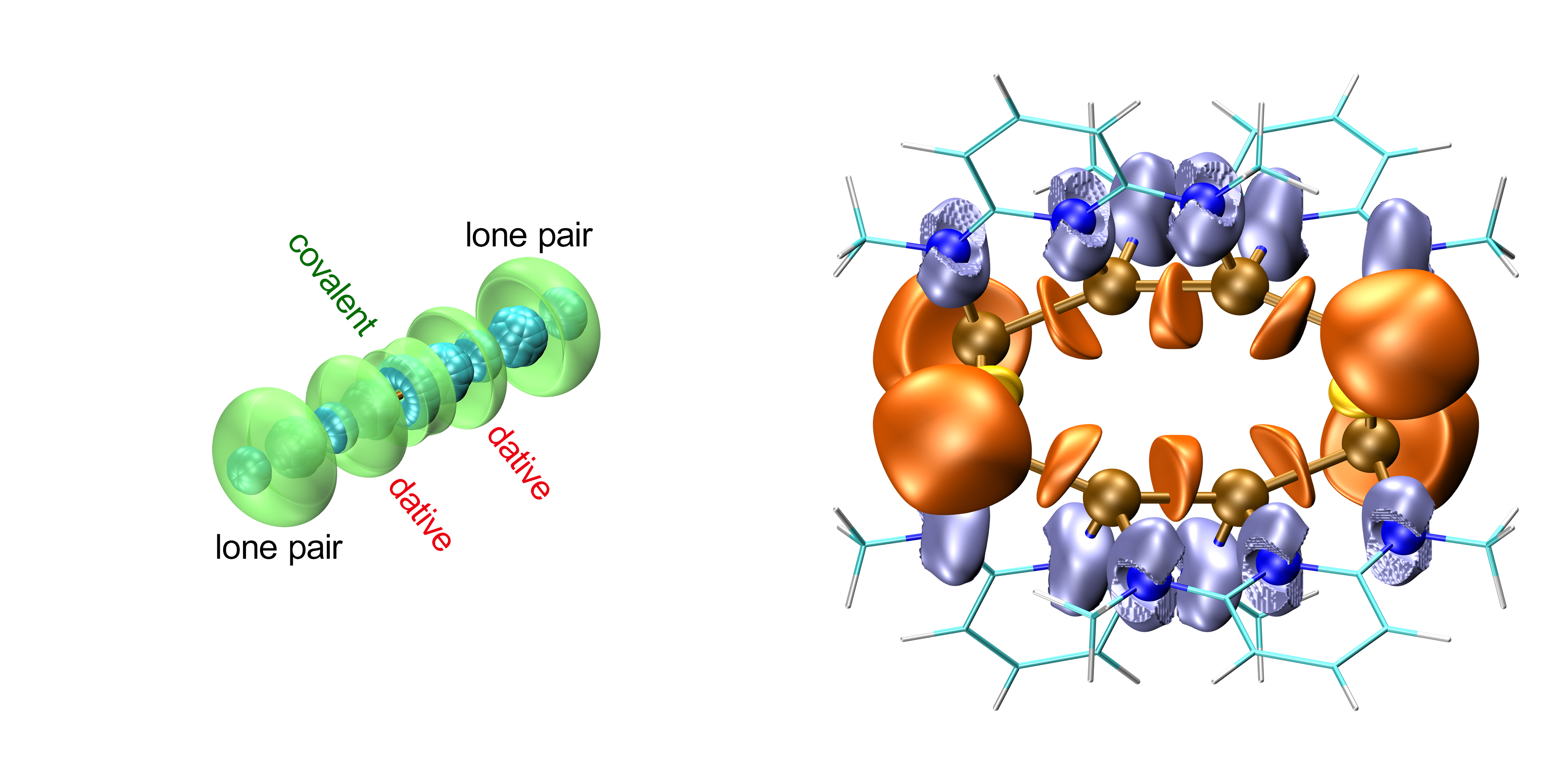
Dative bonds are commonly observed between atoms with different electronegativities. However, the AIM and ELF analysis of the electron density in germanium polycations at the B3LYP/6-31G(d,p) level shows conclusively that geometric and electronic constraints can enforce homonuclear dative bonds.
The dative bonds can be formally described as the interaction of a lone pair of a Ge ion in the center of the cluster with the overall positive charge of another Ge ion. At first glance, the dative bonds resemble weak covalent bonds, but can be readily distinguished from covalent bonds by the properties of the associated bond critical points in the AIM analysis and the shape of their ELF basins. The receiving Ge ion effectively draws electron density from central ions and the associated distribution of positive charge is stabilized by bracketing ligand anions.
The concept of homonuclear dative bonds can be used to rationalize counterintuitive charge pattern in polycations and offers a facile way to predict the disproportion of the germanium backbone of the cluster.